![]()
A Risk Assessment Model for Establishing
Upper Intake Levels for Nutrients
![]()
Food and Nutrition Board
Institute of Medicine
National Academy of Sciences
Washington, DC
June 1998
A Risk Assessment Model for Establishing Upper Intake Levels for Nutrients Food and Nutrition Board Institute of Medicine National Academy of Sciences Washington, DC June 1998![]()
![]()
This is a compilation of the Risk Assessment Model developed in conjunction with the Dietary Reference Intakes activity under the Food and Nutrition Board of the Institute of Medicine, National Academy of Sciences, in the United States.
The Food and Nutrition Board Standing Committee on the Scientific Evaluation of Dietary Reference Intakes is chaired by Vernon R. Young (Massachusetts Institute of Technology); cochaired by John W. Erdman, Jr. (University of Illinois at Urbana-Champaign) and Janet C. King (USDA Western Human Nutrition Research Center and the University of California, Berkeley and Davis); and includes Lindsay H. Allen (University of California, Davis), Stephanie A. Atkinson (McMaster University, Ontario, Canada), Johanna T. Dwyer (New England Medical Center, Boston), John D. Fernstrom (University of Pittsburgh School of Medicine), Scott M. Grundy (University of Texas Southwestern Medical Center at Dallas), Charles H. Hennekens, (Harvard Medical School, Boston), and Sanford A. Miller (University of Texas Health Science Center at San Antonio). The liaison from the DRI Project Steering Committee of the U.S. Interagency Human Nutrition Research Council is Linda Meyers, DHHS Office of Disease Prevention and Health Promotion, and from Health Canada, Peter Fischer. The senior staff for the Institute of Medicine (IOM) include Allison A. Yates, Project Director, and Sandra A. Schlicker, and Carol W. Suitor.
The Subcommittee on Upper Reference Levels of Nutrients is chaired by Ian C. Munro (CanTox, Inc. Ontario, Canada); and includes Walter Mertz (retired, USDA, Beltsville, MD), Rita B. Messing, (Minnesota Department of Health), Sanford A. Miller (University of Texas Health Science Center at San Antonio), Suzanne P. Murphy (University of California, Berkeley and Davis); Joseph V. Rodricks, (ENVIRON Corporation, Arlington, VA), Irwin H. Rosenberg (USDA Human Nutrition Research Center and Tufts University, Boston), Stephen L. Taylor (University of Nebraska, Lincoln), and Robert H. Wasserman (Cornell University, Ithaca, NY). Liaison from Health Canada: Sheila Dubois. The study director is Sandra A. Schlicker.
The Panel on Calcium and Related Nutrients was chaired by Stephanie A. Atkinson (McMaster University, Ontario, Canada); and included Steven A. Abrams (USDA Children’s Nutrition Research Center, Baylor College of Medicine, Houston), Bess Dawson-Hughes (USDA Human Nutrition Research Center on Aging, Tufts University, Boston), Robert P. Heaney (Creighton University, Omaha, Nebraska), Michael F. Holick (Boston University Medical Center), Suzanne P. Murphy (University of California, Berkeley and Davis), Robert K. Rude (University of Southern California, Los Angeles), Bonny L. Specker (South Dakota State University, Brookings), Connie M. Weaver (Purdue University, West Lafayette), and Gary M. Whitford (Medical College of Georgia, Augusta). The study director was Sandra A. Schlicker.
The Panel on Folate, other B Vitamins, and Choline was chaired by Roy M. Pitkin, (Univ. of California, Los Angeles), and included Lindsay H. Allen (University of California, Davis), Merton Bernfield (Harvard Medical School, Boston), Phillipe DeWals (University of Sherbrooke, Quebec, Canada), Ralph Green (University of California, Davis), Donald B. McCormick (Emory University, Atlanta), Robert M. Russell (USDA Human Nutrition Research Center on Aging, Tufts University, Boston), Barry Shane (University of California, Berkeley), an Steven Zeisel (University of North Carolina, Chapel Hill). The study director was Carol W. Suitor.
This project was funded by the U.S. Department of Health and Human Services Office of Disease Prevention and Health Promotion, the National Institutes of Health Office of Nutrition Supplements, the Centers for Disease Control and Prevention, the Food and Drug Administration, the U. S. Department of Agriculture, Agricultural Research Service, the Department of Defense, Health Canada, the Institute of Medicine; and the Dietary Reference Intakes Corporate Donors Fund. Contributors to the Fund to date include Roche Vitamins Inc., Mead Johnson Nutrition Group, Daiichi Fine Chemicals Inc., Kemin Foods, M&M Mars, Weider Nutrition Group, and Natural Source Vitamin E Association.
Copyright© 1998, National Academy of Sciences. All rights reserved.
A Risk Assessment Model for Establishing
Upper Intake Levels for Nutrients
![]()
CONTENTS
Introduction 1
What are Dietary Reference Intakes? 1
Approach for Setting Dietary Reference Intakes, Including Tolerable
Upper Intake Levels 4
Model for the Derivation of Tolerable Upper Intake Levels 5
Risk Assessment and Food Safety 6
Application of the Risk Assessment Model to Nutrients 9
Steps in the Development of the Tolerable Upper Intake Level 13
Derivation of ULs: Summary of Progress to Date 18
References 21
__________
Appendix I: Recommended Dietary Intakes for Individuals 23
Appendix II: Options for Dealing with Uncertainties 25
Appendix III: Reference Heights and Weights for Children and Adults 29
Appendix IV: Case Studies of Application of Model for Risk Assessment
of Upper Levels of Nutrients
A. Calcium 30
B. Folate 42
C. Riboflavin 54
A Risk Assessment Model for Establishing
Upper Intake Levels for Nutrients
![]()
INTRODUCTION
The model for risk assessment of nutrients used to develop tolerable upper intake levels (ULs) is one of the key elements of the developing framework for Dietary Reference Intakes, which are dietary reference values for the intake of nutrients and food components by Americans and Canadians recently released by the U.S. National Academy of Sciences in a series of reports (IOM, 1997; 1998). The overall project is a comprehensive effort undertaken by the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes (DRI Committee) of the Food and Nutrition Board (FNB), Institute of Medicine, National Academy of Sciences in the United States, with active involvement of Health Canada. The DRI project is the result of significant discussion from 1991 to l996 by the FNB regarding how to approach the growing concern that one set of quantitative estimates of recommended intakes, the Recommended Dietary Allowances (RDAs), was scientifically inappropriate to be used as the basis for many of the uses to which they had come to be applied. The lack of specific determinations of maximum or tolerable upper levels of intake was noted (IOM, 1994).
The two DRI reports issued to date provide recommended intakes (see Appendix I) and upper levels of intake for two groups of nutrients and food components: calcium and related nutrients, and folate, B vitamins, and choline. Currently, the DRI Committee and a panel of experts are reviewing dietary antioxidants and related compounds, with similar reviews planned for other micronutrients including trace elements and vitamins A and K, electrolytes and fluid, macronutrients, and other food components not traditionally classified as "nutrients", but purported to play a beneficial role in human diets. It is expected that when evaluation of all groups of nutrients and food components are completed as part of this ongoing process, the model will have been fully developed and validated.
WHAT ARE DIETARY REFERENCE INTAKES?
Dietary Reference Intakes (DRIs) are reference values that are quantitative estimates of nutrient intakes to be used for planning and assessing diets for healthy people. They include both recommended intakes and ULs as reference values (see Figure 1). Although the reference values
|
Type of Use |
For the Individual |
For a Group |
|
|
Planning |
RDA : aim for this intake. |
EAR : use in conjunction with a measure of variability of the group’s intake to set goals for the mean intake of a specific population. |
|
|
AI : aim for this intake. |
|||
|
UL : Use as a guide to limit intake; chronic intake of higher amounts may increase risk of adverse effects. |
|||
|
Assessmenta |
EAR : use to examine the possibility of inadequacy; evaluation of true status requires clinical, biochemical, and/or anthropometric data.UL: use to examine the possibility of overconsumption; evaluation of true status requires clinical, biochemical, and/or anthropometric data. |
EAR : use in the assessment of the preva-lence of inadequate intakes within a group.
|
|
|
RDA = Recommended Dietary Allowance EAR = Estimated Average Requirement AI = Adequate Intake UL = Tolerable Upper Intake Level a Requires statistically valid approximation of usual intake. |
|||
|
Figure 1. Uses of Dietary Reference Intakes for Healthy Individuals and Groups |
|||
are based on data, the data are often scanty or drawn from studies that had limitations in addressing the question. Thus, scientific judgment is required in setting the reference values:
The development of DRIs expands on the periodic reports called Recommended Dietary Allowances, which have been published since 1941 by the National Academy of Sciences.
Recommended Dietary Allowance
The Recommended Dietary Allowance (RDA) is the average daily dietary intake level that is sufficient to meet the nutrient requirement of nearly all (97 to 98 percent) healthy individuals in a particular life-stage (life-stage considers age and, when applicable, pregnancy or lactation) and gender group.
Process for Setting the RDA
The process for setting the RDA depends on being able to set an Estimated Average Requirement (EAR). That is, the RDA is derived from the nutrient requirement; and if an EAR cannot be set, no RDA will be set. The EAR is the daily intake value of a nutrient that is estimated to meet the nutrient requirement of half the healthy individuals in a life-stage and gender group. Before setting the EAR, a specific criterion of adequacy is selected, based on a careful review of the literature. When selecting the criterion, reduction of disease risk is considered along with many other health parameters. The RDA is set at the EAR plus twice the standard deviation (SD) if known (RDA = EAR + 2 SD); if data about variability in requirements are insufficient to calculate an SD, a coefficient of variation for the EAR of 10 percent is ordinarily assumed (RDA = 1.2 × EAR).
The RDA for a nutrient is a value to be used as a goal for dietary intake by healthy individuals. The RDA is not intended to be used to assess the diets of either individuals or groups or to plan diets for groups.
Adequate Intake
The Adequate Intake (AI) is set instead of an RDA if sufficient scientific evidence is not available to calculate an EAR. The AI is based on observed or experimentally-determined estimates of nutrient intake by a group (or groups) of healthy people. For example, the AI for young infants, for whom human milk is the recommended sole source of food for the first 4 to 6 months, is based on the daily mean nutrient intake supplied by human milk for healthy, full-term infants who are exclusively breastfed. The main intended use of the AI is as a goal for the nutrient intake of individuals. Other uses of AIs will be considered by another expert group.
Tolerable Upper Intake Level
The Tolerable Upper Intake Level (UL) is the highest level of daily nutrient intake that is likely to pose no risks of adverse health effects to almost all individuals in the general population. As intake increases above the UL, the risk of adverse effects increases. The term tolerable intake was chosen to avoid implying a possible beneficial effect. Instead, the term is intended to connote a level of intake that can, with high probability, be tolerated biologically. The UL is not intended to be a recommended level of intake. There is no established benefit for healthy individuals if they consume nutrient intakes above the RDA or AI.
ULs are useful because of the increased interest in and availability of fortified foods and the increased use of dietary supplements. ULs are based on total intake of a nutrient from food, water, and supplements if adverse effects have been associated with total intake. However, if adverse effects have been associated with intake from supplements or food fortificants only, the UL is based on nutrient intake from those sources only, not on total intake. The UL applies to chronic daily use.
For many nutrients, there are insufficient data on which to develop a UL. This does not mean that there is no potential for adverse effects resulting from high intake. When data about adverse effects are extremely limited, extra caution may be warranted.
APPROACH FOR SETTING DIETARY REFERENCE INTAKES,
INCLUDING TOLERABLE UPPER INTAKE LEVELS
The scientific data used to develop recommended intakes and ULs have come from observational and experimental studies. Studies published in peer-reviewed journals were the principal source of data. Life stage and gender were considered to the extent possible; but for some nutrients, the data did not provide a basis for proposing different requirements or upper levels for men and women or for adults in different age groups.
Three of the categories of reference values (EAR, RDA, and AI) are defined by specific criteria of nutrient adequacy; the fourth (UL) is defined by a specific end point of adverse effect if one is available. In all cases, data were examined closely to determine whether reduction of risk of a chronic degenerative disease or developmental abnormality could be used as a criterion of adequacy. The quality of studies was examined, considering study design, methods used for measuring intake and indicators of adequacy, and biases, interactions, and confounding factors. After careful review and analysis of the evidence, including examination of the extent of congruence of findings, scientific judgment was used to determine the basis for establishing the values.
Terminology
The term "tolerable" is chosen because it connotes a level of intake that can, with high probability, be tolerated biologically by individuals; it does not imply acceptability of that level in any other sense. The setting of a UL does not indicate that nutrient intakes greater than the RDA or AI are recommended as being beneficial to an individual. Many individuals are self-medicating with nutrients for curative or treatment purposes. It is beyond the scope of the model at this time to address the possible therapeutic benefits of higher nutrient intakes that may offset the risk of adverse effects. The UL is not meant to apply to individuals who are treated with the nutrient or food component under medical supervision.
The term adverse effect is defined as any significant alteration in the structure or function of the human organism (Klaassen et al., 1986), or any impairment of a physiologically important function, in accordance with the definition set by the joint World Health Organization, Food and Agriculture Organization of the United Nations, and International Atomic Energy Agency (WHO/FAO/IAEA) Expert Consultation in Trace Elements in Human Nutrition and Health (WHO, 1996). In the case of nutrients, it is exceedingly important to consider the possibility that the intake of one nutrient may alter in detrimental ways the health benefits conferred by another nutrient. Any such alteration (referred to as an adverse nutrient-nutrient interaction) is considered an adverse health effect. When evidence for such adverse interactions is available, it is considered in establishing a nutrient’s UL.
Concept
Like all chemical agents, nutrients can produce adverse health effects if intakes from any combination of food, water, nutrient supplements, and pharmacologic agents is excessive. Some lower level of nutrient intake will ordinarily pose no likelihood (or risk) of adverse health effects in normal individuals even if the level is above that associated with any benefit. It is not possible to identify a single "risk-free" intake level for a nutrient that can be applied with certainty to all members of a population. However, it is possible to develop intake levels that are unlikely to pose risks of adverse health effects to most members of the general population, including sensitive individuals. For some nutrients or food components these intake levels may, however, pose a risk to subpopulations with extreme or distinct vulnerabilities.
MODEL FOR THE DERIVATION OF TOLERABLE
UPPER INTAKE LEVELS
The possibility that the methodology used to derive ULs might be reduced to a mathematical model that could be generically applied to all nutrients was considered. Such a model might have several potential advantages, including ease of application and assurance of consistent treatment of all nutrients. It was concluded, however, that the current state of scientific understanding of toxic phenomena in general, and nutrient toxicity in particular, is insufficient to support the development of such a model. Scientific information regarding various adverse effects and their relationships to intake levels varies greatly among nutrients and depends on the nature, comprehensiveness, and quality of available data. The uncertainties associated with the unavoidable problem of extrapolating from the circumstances under which data are developed (for example, in the laboratory or clinic) to other circumstances (for example, to the healthy population) adds to the complexity.
Given the current state of knowledge, any attempt to capture in a mathematical model all the information and scientific judgments that must be made to reach conclusions regarding ULs would not be consistent with contemporary risk assessment practices. Instead, the model for the derivation of ULs consists of a set of scientific factors that always should be considered explicitly. The framework under which these factors are organized is called risk assessment. Risk assessment (NRC, 1983, 1994) is a systematic means of evaluating the probability of occurrence of adverse health effects in humans from excess exposure to an environmental agent (in this case, a nutrient or food component) (FAO/WHO, 1995; Health Canada, 1993). The hallmark of risk assessment is the requirement to be explicit in all the evaluations and judgments that must be made to document conclusions.
RISK ASSESSMENT AND FOOD SAFETY
Basic Concepts
Risk assessment is a scientific undertaking having as its objective a characterization of the nature and likelihood of harm resulting from human exposure to agents in the environment. The characterization of risk typically contains both qualitative and quantitative information and includes a discussion of the scientific uncertainties in that information. In the present context, the agents of interest are nutrients, and the environmental media are food, water, and nonfood sources such as nutrient supplements and pharmacologic preparations.
Performing a risk assessment results in a characterization of the relationships between exposure(s) to an agent and the likelihood that adverse health effects will occur in members of exposed populations. Scientific uncertainties are an inherent part of the risk assessment process and are discussed below. Deciding whether the magnitude of exposure is "acceptable" in specific circumstances is not a component of risk assessment; this activity falls within the domain of risk management. Risk management decisions depend on the results of risk assessments but may also involve the public health significance of the risk, the technical feasibility of achieving various degrees of risk control, and the economic and social costs of this control. Because there is no single, scientifically definable distinction between "safe" and "unsafe" exposures, risk management necessarily incorporates components of sound, practical decision making that are not addressed by the risk assessment process (NRC, 1983, 1994).
A risk assessment requires that information be organized in rather specific ways but does not require any specific scientific evaluation methods. Rather, risk assessors must evaluate scientific information using what they judge to be appropriate methods; and they must make explicit the basis for their judgments, the uncertainties in risk estimates, and when appropriate, alternative interpretations of the available data that may be scientifically plausible (NRC, 1994; OTA, 1993).
Risk assessment is subject to two types of scientific uncertainties: (1) those related to data and (2) those associated with inferences that are required when directly applicable data are not available (NRC, 1994). Data uncertainties arise when evaluating information obtained from the epidemiologic and toxicologic studies of nutrient intake levels that are the basis for risk assessments. Examples of inferences include the use of data from experimental animals to estimate responses in humans and the selection of uncertainty factors to estimate inter- and intraspecies variabilities in response to toxic substances. Uncertainties arise whenever estimates of adverse health effects in humans are based on extrapolations of data obtained under dissimilar conditions (for example, from experimental animal studies). Options for dealing with uncertainties are discussed below and in detail in Appendix II.
Steps in the Risk Assessment Process
The organization of risk assessment is based on a model proposed by the NRC (1983, 1994); that model is widely used in public health and regulatory decision making. The steps of risk assessment as applied to nutrients are as follows (see also Figure 2):
• Step 1. Hazard identification involves the collection, organization, and evaluation of all information pertaining to the adverse effects of a given nutrient. It concludes with a summary of the evidence concerning the capacity of the nutrient to cause one or more types of toxicity in humans.
• Step 2. Dose-response assessment determines the relationship between nutrient intake (dose) and adverse effect (in terms of incidence and severity). This step concludes with an estimate of the UL—it identifies the highest level of daily nutrient intake that is likely to pose no risks of adverse health effects to almost all individuals in the general population. Different ULs may be developed for various life-stage groups.
• Step 3. Intake assessment evaluates the distribution of usual total daily nutrient intakes among members of the general population.
• Step 4. Risk characterization summarizes the conclusions from Steps 1 through 3 and evaluates the risk. Generally, the risk is expressed as the fraction of the exposed population, if any, having nutrient intakes (Step 3) in excess of the estimated UL (Steps 1 and 2). If possible,
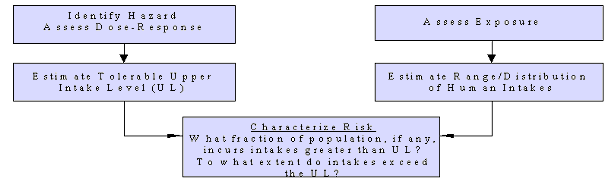
Figure 2. Risk Assessment Model for Nutrient Toxicity
characterization also covers the magnitude of any such excesses. Scientific uncertainties associated with both the UL and the intake estimates are described so that risk managers understand the degree of scientific confidence they can place in the risk assessment.
The risk assessment contains no discussion of recommendations for reducing risk; these are the focus of risk management.
Thresholds
A principal feature of the risk assessment process for noncarcinogens is the long-standing acceptance that no risk of adverse effects is expected unless a threshold dose (or intake) is exceeded. The adverse effects that may be caused by a nutrient or food component almost certainly occur only when the threshold dose is exceeded (NRC, 1994; WHO, 1996). The critical issues concern the methods used to identify the approximate threshold of toxicity for a large and diverse human population. Because most nutrients are not considered to be carcinogenic in humans, the approach to carcinogenic risk assessment (EPA, 1996) is not discussed here.
Thresholds vary among members of the general population (NRC, 1994). For any given adverse effect, if the distribution of thresholds in the population could be quantitatively identified, it would be possible to establish ULs by defining some point in the lower tail of the distribution of thresholds that would be protective for some specified fraction of the population. However, data are not sufficient to allow identification of the distribution of thresholds for all but a few, well-studied nutrients and compounds found in food (for example, acute toxic effects or for chemicals such as lead, where the human database is very large). The method for identifying thresholds for a general population described here is designed to ensure that almost all members of the population will be protected, but it is not based on an analysis of the theoretical (but practically unattainable) distribution of thresholds. By using the model to derive the threshold, however, there is considerable confidence that the threshold, which becomes the UL for nutrients or food components, lies very near the low end of the theoretical distribution, and is the end representing the most sensitive members of the population. For some nutrients, there may be subpopulations that are not included in the general distribution because of extreme or distinct vulnerabilities to toxicity. Such distinct groups, whose conditions warrant medical supervision, may not be protected by the UL.
The Joint FAO/WHO Expert Commission on Food Additives and various national regulatory bodies have identified factors (called uncertainty factors) that account for interspecies and intraspecies differences in response to the hazardous effects of substances and to account for other uncertainties (WHO, 1987). These factors are used to make inferences about the threshold dose of substances for members of a large and diverse human population from data on adverse effects obtained in epidemiological or experimental studies. These factors are applied consistently when data of specific types and quality are available. They are typically used to derive acceptable daily intakes for food additives and other substances for which data on adverse effects are considered sufficient to meet minimum standards of quality and completeness (FAO/WHO, 1982). These adopted or recognized uncertainty factors have sometimes been coupled with other factors to compensate for deficiencies in the available data and other uncertainties regarding data.
When possible, the UL is based on a no-observed-adverse-effect level (NOAEL), which is the highest intake (or experimental oral dose) of a nutrient at which no adverse effects have been observed in the individuals studied. This is identified for a specific circumstance in the hazard identification and dose-response assessment steps of the risk assessment. If there are no adequate data demonstrating a NOAEL, then a lowest-observed-adverse-effect level (LOAEL) may be used. A LOAEL is the lowest intake (or experimental oral dose) at which an adverse effect has been identified. The derivation of a UL from a NOAEL (or LOAEL) involves a series of choices about what factors should be used to deal with uncertainties. Uncertainty factors (UFs) are applied in an attempt both to deal with gaps in data and with incomplete knowledge regarding the inferences required (for example, the expected variability in response within the human population). The problems of both data and inference uncertainties arise in all steps of the risk assessment. A discussion of options available for dealing with these uncertainties is presented below and in greater detail in the Appendix II.
A UL is not, in itself, a description of human risk. It is derived by application of the hazard identification and dose-response evaluation steps (Steps 1 and 2) of the risk assessment model. To determine whether populations are at risk requires an intake or exposure assessment (Step 3, evaluation of their intakes of the nutrient) and a determination of the fractions of those populations, if any, whose intakes exceed the UL. In the intake assessment and risk characterization steps (Steps 3 and 4), the distribution of actual intakes for the population is used as a basis in determining whether and to what extent the population is at risk.
APPLICATION OF THE RISK ASSESSMENT MODEL
TO NUTRIENTS
This section provides guidance for applying the risk assessment framework (the model) to the derivation of ULs for nutrients.
Special Problems Associated with Substances Required
for Human Nutrition
Although the risk assessment model outlined above can be applied to nutrients to derive ULs, it must be recognized that nutrients possess some properties that distinguish them from the types of agents for which the risk assessment model has originally developed (NRC, 1983). In the application of accepted standards for assessing risks of environmental chemicals to the risk assessment of nutrients and food components, a fundamental difference between the two categories must be recognized: within a certain range of intakes, many nutrients are essential for human well-being and usually for life itself. Nonetheless, they may share with other chemicals the production of adverse effects at excessive exposures. Because the consumption of balanced diets is consistent with the development and survival of humankind over many millennia, there is less need for the large uncertainty factors that have been used in the typical risk assessment of nonessential chemicals. In addition, if data on the adverse effects of nutrients are available primarily from studies in human populations, there will be less uncertainty than is associated with the types of data available on nonessential chemicals.
There is no evidence to suggest that nutrients consumed at the recommended intake (the RDA or AI) present a risk of adverse effects to the general population. It is clear, however, that the addition of nutrients to a diet, either through the ingestion of large amounts of highly fortified food or nonfood sources such as supplements, or both, may (at some level) pose a risk of adverse health effects. The UL is the highest level of daily nutrient intake that is likely to pose no risks of adverse health effects to almost all individuals in the general population. As intake increases above the UL, the risk of adverse effects increases.
If adverse effects have been associated with total intake, ULs are based on total intake of a nutrient from food, water, and supplements. For cases in which adverse effects have been associated with intake only from supplements and/or food fortificants, the UL is based on intake from those sources only, rather than on total intake. The effects of nutrients from fortified foods or supplements may differ from those of naturally occurring constituents of foods because of several factors: the chemical form of the nutrient, the timing of the intake and amount consumed in a single bolus dose, the matrix supplied by the food, and the relation of the nutrient to the other constituents of the diet. Nutrient requirements and food intake are related to the metabolizing body mass, which is also at least an indirect measure of the space in which the nutrients are distributed. This relation between food intake and space of distribution supports homeostasis, which maintains nutrient concentrations in that space within a range compatible with health. However, excessive intake of a single nutrient from supplements or fortificants may compromise this homeostatic mechanism. Such elevations alone may pose risks of adverse effects; imbalances among the concentrations of mineral elements (for example, calcium, iron, zinc, an copper) can result in additional risks (Mertz et al., 1994). These reasons and those discussed previously support the need to include the form and pattern of consumption in the assessment of risk from high nutrient intake.
Consideration of Variability in Sensitivity
This risk assessment model outlined in this paper is consistent with classical risk assessment approaches in that it must consider variability in the sensitivity of individuals to adverse effects of nutrients. A discussion of how variability is dealt with in the context of nutritional risk assessment follows.
Physiological changes and common conditions associated with growth and maturation that occur during an individual’s lifespan may influence sensitivity to nutrient toxicity. For example, (1) sensitivity increases with declines in lean body mass and with declines in renal and liver function that occur with aging; (2) sensitivity changes in direct relation to intestinal absorption or intestinal synthesis of nutrients (for example, Vitamin K, biotin); (3) in the newborn infant, sensitivity is also increased because of rapid brain growth and limited ability to secrete or biotransform toxicants; and (4) sensitivity increases with decreases in the rate of metabolism of nutrients. During pregnancy, the increase in total body water and glomerular filtration results in lower blood levels of water soluble vitamins dose-for-dose, and therefore, reduced susceptibility to potential adverse effects. However, in the unborn fetus this may be offset by active placental transfer, accumulation of certain nutrients in the amniotic fluid, and rapid development of the brain. Examples of life-stage groups that may differ in terms of nutritional needs and toxicological sensitivity include infants and children, the elderly population, and women during pregnancy and lactation.
Even within relatively homogeneous life-stage groups, there is a range of sensitivities to toxic effects. The model described below accounts for normally expected variability in sensitivity, but it excludes subpopulations with extreme and distinct vulnerabilities. Such subpopulations consist of individuals needing medical supervision; they are better served through the use of public health screening, product labeling, or other individualized health care strategies. (Such populations may not be at "negligible risk" when their intakes reach the UL developed for the healthy population.) The decision to treat identifiable vulnerable subgroups as distinct (not protected by the UL) is a matter of judgment and is made evident in the rationale provided for characterizing the UL.
Bioavailability
In the context of toxicity, the bioavailability of an ingested nutrient can be defined as its accessibility to normal metabolic and physiological processes. Bioavailability influences a nutrient’s beneficial effects at physiological levels of intake and also may affect the nature and severity of toxicity due to excessive intakes. Factors that affect bioavailability include the concentration and chemical form of the nutrient, the nutrition and health of the individual, and excretory losses. Bioavailability data for specific nutrients must be considered and incorporated by the risk assessment process.
Some nutrients, such as folate, may be less readily absorbed when they are part of a meal than when taken separately. Supplemental forms of some nutrients, such as some of the B vitamins, phosphorus, or magnesium, may require special consideration if they have higher bioavailability and therefore may present a higher risk of producing adverse effects than equivalent amounts from the natural form found in food.
Nutrient-Nutrient Interactions
A diverse array of adverse health effects can occur as a result of the interaction of nutrients. The potential risks of adverse nutrient-nutrient interactions increase when there is an imbalance in the intake of two or more nutrients. Excessive intake of one nutrient may interfere with absorption, excretion, transport, storage, function, or metabolism of a second nutrient. For example, dietary interactions can affect the chemical forms of elements at the site of absorption through ligand binding or changes in the valence state of an element (Mertz et al., 1994). Phytates, phosphates, and tannins are among the most powerful depressants of bioavailability, and organic acids, such as citric and ascorbic acid, are strong enhancers for some minerals and trace elements. Thus dietary interactions strongly influence the bioavailability of elements by affecting their partition between the absorbed and the nonabsorbed portion of the diet. The large differences of bioavailability ensuing from these interactions support the need to specify the chemical form of the nutrient when setting ULs. Dietary interactions can also alter nutrient bioavailability through their effect on excretion. For example, dietary intake of protein, phosphorus, sodium, and chloride all affect urinary calcium excretion and hence calcium bioavailability. Interactions that significantly elevate or reduce bioavailability may represent adverse health effects.
Although it is critical to include knowledge of any such interactions in the risk assessment, it is difficult to evaluate the possibility of interactions without reference to a particular level of intake. This difficulty can be overcome if a UL for a nutrient or food component is first derived based on other measures of toxicity. Then an evaluation can be made of whether intake at the UL has the potential to affect the bioavailability of other nutrients.
Possible adverse nutrient-nutrient interactions, then, are considered as a part of setting a UL. Nutrient-nutrient interactions may be considered either as a critical endpoint on which to base a UL for that nutrient or as supportive evidence for a UL based on another endpoint.
Other Relevant Factors Affecting Bioavailability of Nutrients
In addition to nutrient interactions, other considerations have the potential to influence nutrient bioavailability, such as the nutritional status of an individual and the form of intake. These issues should be considered in the risk assessment. The absorption and utilization of most minerals, trace elements, and some vitamins are a function of the individual’s nutritional status, particularly regarding the intake of other specific nutrients such as iron (Barger-Lux et al., 1995; Mertz et al., 1994).
With regard to the form of intake, minerals and trace elements often are less readily absorbed when they are part of a meal than when taken separately or when present in drinking water (NRC, 1989). The opposite is true for fat-soluble vitamins whose absorption depends on fat in the diet. ULs must therefore be based on nutrients as part of the total diet, including the contribution from water. Nutrient supplements that are taken separately from food require special consideration, since they are likely to have different availabilities and therefore may represent a greater risk of producing toxic effects.
STEPS IN THE DEVELOPMENT OF THE TOLERABLE
UPPER INTAKE LEVEL
Step 1. Hazard Identification
Based on a thorough review of the scientific literature, the hazard identification step outlines the adverse health effects that have been demonstrated to be caused by the nutrient. The primary types of data used as background for identifying nutrient hazards in humans are as follows:
• Human studies. Human data provide the most relevant kind of information for hazard identification, and, when they are of sufficient quality and extent, are given greatest weight. However, the number of controlled human toxicity studies conducted in a clinical setting is very limited for ethical reasons. Such studies are generally most useful for identifying very mild (and ordinarily reversible) adverse effects. Observational studies that focus on well-defined populations with clear exposures to a range of nutrient intake levels are useful for establishing a relationship between exposure and effect. Observational data in the form of case reports or anecdotal evidence are used for developing hypotheses that can lead to knowledge of causal associations. Sometimes a series of case reports, if it shows a clear and distinct pattern of effects, may be reasonably convincing on the question of causality.
• Animal studies. The majority of the available data used in regulatory risk assessments comes from controlled laboratory experiments in animals, usually mammalian species other than humans (for example, rodents). Such data are used in part because human data on food derived substances, particularly nonessential chemicals, are generally very limited. Because well-conducted animal studies can be controlled, establishing causal relationships is generally not difficult. However, cross species differences make the usefulness of animal data for establishing ULs problematic (see below).
Six key issues that are addressed in the data evaluation of human and animal studies are the following:
1. Evidence of adverse effects in humans. The hazard identification step involves the examination of human, animal, and in vitro published evidence addressing the likelihood of a nutrient or food component eliciting an adverse effect in humans. Decisions regarding which observed effects are adverse are based on scientific judgments. Although toxicologists generally regard any demonstrable structural or functional alteration to represent an adverse effect, some alterations may be considered of little or self-limiting biological importance. As noted earlier, adverse nutrient-nutrient interactions are considered in the definition of an adverse effect.
2. Causality. Is a causal relationship established by the published human data? The criteria of Hill (1971) are considered in judging the causal significance of an exposure-effect association indicated by epidemiologic studies. These criteria include: demonstration of a temporal relationship, consistency, narrow confidence intervals for risk estimates, a biological gradient or dose response, specificity of effect, biological plausibility, and coherence.
3. Relevance of experimental data. Consideration of the following issues can be useful in assessing the relevance of experimental data.
• Animal data. Animal data may be of limited utility in judging the toxicity of nutrients because of highly variable interspecies differences in nutrient requirements. Nevertheless, relevant animal data are considered in the hazard identification and dose-response assessment steps where applicable.
• Route of exposure. Data derived from studies involving oral exposure (rather than parenteral, inhalation, or dermal exposure) are most useful for the evaluation of nutrients and food components. Data derived from studies involving parenteral, inhalation, or dermal routes of exposure may be considered relevant if the adverse effects are systemic and data are available to permit interroute extrapolation.
• Duration of exposure. Because the magnitude, duration, and frequency of exposure can vary considerably in different situations, consideration needs to be given to the relevance of the exposure scenario (for example, chronic daily dietary exposure versus short-term bolus doses) to dietary intakes by human populations.
4. Mechanisms of toxic action. Knowledge of molecular and cellular events underlying the production of toxicity can assist in dealing with the problems of extrapolation between species and from high to low doses. It may also aid in understanding whether the mechanisms associated with toxicity are those associated with deficiency. In most cases, however, because knowledge of the biochemical sequence of events resulting from toxicity and deficiency is still incomplete, it is not yet possible to state with certainty whether or not these sequences share a common pathway. Iron, the most thoroughly studied trace element, may represent the only exception to this statement. Deficient to near-toxic exposures share the same pathway, which maintains controlled oxygen transport and catalysis. Toxicity sets in when the exposure exceeds the specific iron-complexing capacity of the organism, resulting in free iron species initiating peroxidation.
5. Quality and completeness of the database. The scientific quality and quantity of the database are evaluated. Human or animal data are reviewed for suggestions that the substances have the potential to produce additional adverse health effects. If suggestions are found, additional studies may be recommended.
6. Identification of distinct and highly sensitive subpopulations. The ULs are based on protecting the most sensitive members of the general population from adverse effects of high nutrient intake. Some highly sensitive subpopulations have responses (in terms of incidence, severity, or both) to the agent of interest that are clearly distinct from the responses expected for the healthy population. The risk assessment process recognizes that there may be individuals within any life-stage group that are more biologically sensitive than others, and thus their extreme sensitivities do not fall within the range of sensitivities expected for the general population. The UL for the general population may not be protective for these subgroups. As indicated earlier, the extent to which a distinct subpopulation will be included in the derivation of a UL for the general population is an area of judgment to be addressed on a case-by-case basis.
Step 2. Dose-Response Assessment
The process for deriving the UL is described in this section and outlined in Figure 3. It includes selection of the critical data set, identification of a critical endpoint with its NOAEL (or LOAEL), and assessment of uncertainty.
Data Selection
The data evaluation process results in the selection of the most appropriate or critical data set(s) for deriving the UL. Selecting the critical data set includes the following considerations:
• Human data are preferable to animal data.
• In the absence of appropriate human data, information from an animal species whose biological responses are most like those of humans is most valuable.
• If it is not possible to identify such a species or to select such data, data from the most sensitive animal species, strain, or gender combination are given the greatest emphasis.
Components Of Hazard Identification
• Evidence of adverse effects in humans
• Causality
• Relevance of experimental data
• Mechanisms of toxic action
• Quality and completeness of the database
• Identification of distinct and highly sensitive subpopulations
Components Of Dose-Response Assessment
• Data selection
• Identification of no-observed-adverse-effect level (NOAEL) (or lowest-observed-adverse-effect level [LOAEL]) and critical endpoint
• Uncertainty assessment
• Derivation of a UL
• Characterization of the estimate and special considerations
Figure 3: Development of Tolerable Upper Intake Levels (ULs)
• The route of exposure that most resembles the route of expected human intake is preferable. This includes considering the digestive state (for example, fed or fasted) of the subjects or experimental animals. Where this is not possible, the differences in route of exposure are noted as a source of uncertainty.
• The critical data set defines a dose-response relationship between intake and the extent of the toxic response known to be most relevant to humans. Data on bioavailability are considered and adjustments in expressions of dose-response are made to determine whether any apparent differences in response can be explained. For example, it is known that different metal salts can display different degrees of bioavailability. If the database involves studies of several different salts (for example, iron or chromium valence states), and the effect of the nutrient is systemic, then apparent differences in the degree and/or form of the toxic response among different salts may simply reflect differences in bioavailability. Data on bioavailability are considered and adjustments in expressions of dose-response are made to determine whether any apparent differences in response can be explained.
• The critical data set documents the route of exposure and the magnitude and duration of the intake. Furthermore, the critical data set documents the intake that does not produce adverse effects (the NOAEL), as well as the intake producing toxicity.
Identification of NOAEL (or LOAEL) and Critical Endpoint
A nutrient can produce more than one toxic effect (or endpoint), even within the same species or in studies using the same or different exposure durations. The NOAELs and (LOAELs) for these effects will differ. The critical endpoint used to establish a UL is the adverse biological effect exhibiting the lowest NOAEL (for example, the most sensitive indicator of a nutrient or food component’s toxicity). The derivation of a UL based on the most sensitive endpoint will ensure protection against all other adverse effects.
For some nutrients, there may be inadequate data on which to develop a UL. The lack of reports of adverse effects following excess intake of a nutrient does not mean that adverse effects do not occur. As the intake of any nutrient increases, a point (A, see Figure 4) is reached at which intake begins to pose a risk. Above this point, increased intake increases the risk of adverse effects. For some nutrients, and for various reasons, there are inadequate data to identify point A, or even to make any estimate of its location.
Because adverse effects are almost certain to occur for any nutrient at some level of intake, it should be assumed that such effects may occur for nutrients for which a scientifically documentable UL cannot now be derived. Until a UL is set or an alternative approach to identifying protective limits is developed, intakes greater than the RDA or AI should be viewed with caution.
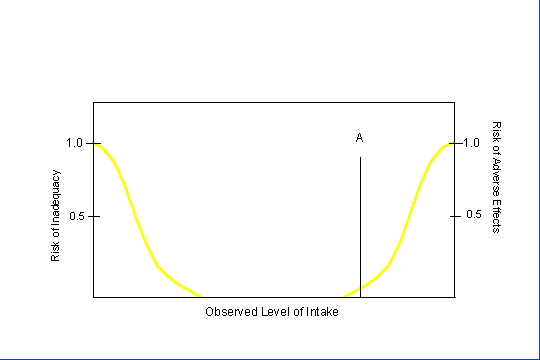
Figure 4. Theoretical Description of Health Effects of a Nutrient as a Function of Level of Intake.
The Tolerable Upper Intake Level (UL) is the highest level of daily nutrient intake that is likely to pose no risks of adverse health effects to almost all individuals in the general population. At intakes above the UL, the risk of adverse effects increases.
Uncertainty Assessment
Several judgments must be made regarding the uncertainties and thus the uncertainty factor (UF) associated with extrapolating from the observed data to the general population (see Appendix II). Applying a UF to a NOAEL (or LOAEL) results in a value for the derived UL that is less than the experimentally derived NOAEL, unless the UF is 1.0. The larger the uncertainty, the larger the UF and the smaller the UL. This is consistent with the ultimate goal of the risk assessment: to provide an estimate of a level of intake that will protect the health of the healthy population (Mertz et al., 1994).
Although several reports describe the underlying basis for UFs (Dourson and Stara, 1983; Zielhuis and van der Kreek, 1979), the strength of the evidence supporting the use of a specific UF will vary. Because the imprecision of these UFs is a major limitation of risk assessment approaches, considerable leeway must be allowed for the application of scientific judgment in making the final determination. Since data are generally available regarding intakes of nutrients and food components in human populations, the data on nutrient toxicity may not be subject to the same uncertainties as with nonessential chemical agents resulting in UFs for nutrients and food components typically less than 10. They are lower with higher quality data and when the adverse effects are extremely mild and reversible.
In general, when determining a UF, the following potential sources of uncertainty are considered and combined into the final UF:
• Interindividual variation in sensitivity. Small UFs (close to 1) are used if it is judged that little population variability is expected for the adverse effect, and larger factors (close to 10) are used if variability is expected to be great (NRC, 1994).
• Experimental animal to human. A UF is generally applied to the NOAEL to account for the uncertainty in extrapolating animal data to humans. Larger UFs (close to 10) may be used if it is believed that the animal responses will underpredict average human responses (NRC, 1994).
• LOAEL to NOAEL. If a NOAEL is not available, a UF may be applied to account for the uncertainty in deriving a UL from the LOAEL. The size of the UF involves scientific judgment based on the severity and incidence of the observed effect at the LOAEL and the steepness (slope) of the dose response.
• Subchronic NOAEL to predict chronic NOAEL. When data are lacking on chronic exposures, scientific judgment is necessary to determine whether chronic exposure is likely to lead to adverse effects at lower intakes than those producing effects after subchronic exposures (exposures of shorter duration).
Characterization of the Estimate and Special Considerations
ULs are derived for various life-stage groups using relevant databases, NOAELs and LOAELs, and UFs. In cases where no data exist with regard to NOAELs or LOAELs for the group under consideration, extrapolations from data in other age groups and/or animal data are made on the basis of known differences in body size, physiology, metabolism, absorption, and excretion of the nutrient.
If the data review reveals the existence of subpopulations having distinct and exceptional sensitivities to a nutrient’s toxicity, these subpopulations should be explicitly discussed and concerns related to adverse effects noted; however the use of the data is not included in the identification of the NOAEL or LOAEL upon which the UL for the general population is based.
DERIVATION OF ULs: SUMMARY OF PROGRESS TO DATE
Derivation of UFs
The model described in this document has been applied to two groups of nutrients and food components as part of the continuing DRI process. The selection of a UF of approximately 1.0 for fluoride and magnesium is primarily based on the very mild (and in the case of magnesium, reversible) nature of the adverse effects observed. A slightly larger UF (1.2) was selected for vitamin D intake in adults and in other life-stage groups except infants as the short duration of the study used (Narang et al., 1984) and the small sample size supports the selection of a slightly larger UF. For vitamin D in infants, a larger UF (1.8) was selected due to the insensitivity of the critical endpoint, the small sample sizes of the studies, and the limited data about the sensitivity at the tails of the distribution. A UF of 2 was selected for calcium to account for the potential increased susceptibility to high calcium intake by individuals who form renal stones and the potential adverse effects in the range between normal phosphorus levels and levels associated with ectopic mineralization. The selection of a UF for phosphorus is due to the relative lack of human data describing adverse effects of excess phosphorus intakes.
With regard to the B vitamins and choline, because of lack of suitable data that met the requirements of the model, NOAELs (and LOAELs) could not be set for thiamin, riboflavin, vitamin B12, pantothenic acid, or biotin. The UF for synthetic folic acid was 5, based primarily on the uncontrolled observation that millions of people to date had been exposed to self treatment with about 0.1 of the LOAEL as 400 m g in vitamin pills without reporting harm. For niacin as a supplement or food fortificant, the UF selected was 1.5, based on the transient nature of the adverse effect of flushing, and the consideration that it was applied to a LOAEL and not a NOAEL. The UF for both vitamin B6 and choline was 2. In the case of vitamin B6, there was less data available involving responses to pyridoxine doses under 500 mg/day, and thus more limited information upon which to base a UL. The UF of 2 for choline was selected because of the limited data regarding hypotension and the magnitude of the interindividual variation in response to cholinergic effects.
Derivation of a UL
UL values have been established for broad age groups for nutrients for which adequate data are available (see Table 1). Values are set at levels that are unlikely to pose risk to the most sensitive members of the general population and cannot be used to assess the prevalence of the population at risk for adverse effects as a result of excess intakes. The UL for magnesium is from supplement intake only, and for niacin and folate (in the form of folic acid) from fortified food and supplement intake only. In all three cases, the nutrient naturally found in foods is excluded from concern. The adverse effect or critical endpoint which is used for each nutrient is given in Table 2. Three case studies (calcium, folate, and biotin) are described in Appendix IV.
Derivation of a UL for Other Groups
The UL is derived by dividing the NOAEL (or LOAEL) by a single UF that incorporates all relevant uncertainties for the lifestage category for which the data is available (see Table 1). The derivation of a UL involves the use of scientific judgment to select the appropriate NOAEL (or LOAEL) and UF. The risk assessment requires explicit consideration and discussion of all choices made, both regarding the data used and the uncertainties accounted for.
For infants, ULs were not determined for any of the B vitamins, choline, magnesium, phosphorus, or calcium because of the lack of data on adverse effects in this age group and concern regarding infants’ possible lack of ability to handle excess amounts. Thus, caution is warranted; food should be the source of intake of these nutrients by infants. For vitamin D and fluoride, due to the significant information on effects from various levels of intake by infants for these nutrients, ULs were developed.
Table 1. Tolerable Upper Intake Levels (ULa) for Certain Nutrients and Food Components
|
Life-Stage Group |
Calcium (g/day) |
Phosphorus (g/day) |
Magnesium (mg/day) b |
Vitamin D (m g/day) |
Fluoride (mg/day) |
Niacin (mg/day)c |
Vitamin B6 (mg/day) |
Synthetic Folic Acid (m g/day)c |
Choline (g/day) |
|
0- 6 months |
NDd |
ND |
ND |
25 |
0.7 |
ND |
ND |
ND |
ND |
|
7- 12 months |
ND |
ND |
ND |
25 |
0.9 |
ND |
ND |
ND |
ND |
|
1- 3 years |
2.5 |
3 |
65 |
50 |
1.3 |
10 |
30 |
300 |
1.0 |
|
4- 8 years |
2.5 |
3 |
110 |
50 |
2.2 |
15 |
40 |
400 |
1.0 |
|
9- 13 years |
2.5 |
4 |
350 |
50 |
10 |
20 |
60 |
600 |
2.0 |
|
14- 18 years |
2.5 |
4 |
350 |
50 |
10 |
30 |
80 |
800 |
3.0 |
|
19- 70 years |
2.5 |
4 |
350 |
50 |
10 |
35 |
100 |
1,000 |
3.5 |
|
> 70 years |
2.5 |
3 |
350 |
50 |
10 |
35 |
100 |
1,000 |
3.5 |
|
Pregnancy |
|||||||||
|
£ 18 years |
2.5 |
3.5 |
350 |
50 |
10 |
30 |
80 |
800 |
3.0 |
|
19- 50 years |
2.5 |
3.5 |
350 |
50 |
10 |
35 |
100 |
1,000 |
3.5 |
|
Lactation |
|||||||||
|
£ 18 years |
2.5 |
4 |
350 |
50 |
10 |
30 |
80 |
800 |
3.0 |
|
19- 50 years |
2.5 |
4 |
350 |
50 |
10 |
35 |
100 |
1,000 |
3.5 |
a
UL = the maximum level of daily nutrient intake that is likely to pose no risk of adverse effects. Unless otherwise specified, the UL represents total intake from food, water, and supplements. Due to lack of suitable data, ULs could not be established for thiamin, riboflavin, vitamin B12, pantothenic acid, or biotin. In the absence of ULs, extra caution may be warranted in consuming levels above recommended intakes.b
The UL for magnesium represents itnake from a pharmacological agent only and does not include intake from food and water.c
The ULs for niacin and synthetic folic acid apply to forms obtained from supplements, fortified foods, or a combination of the two.d
ND: Not determinable due to lack of data of adverse effects in this age group and concern with regard to lack of ability to handle excess amounts. Source of intake should be from food only to prevent high levels of intake.
Table 2. UL Critical Adverse Effects
|
Nutrient |
Adverse Effect |
|
Calcium |
Milk-alkali syndrome |
|
Phosphorus |
Elevated serum Pi |
|
Magnesium |
Osmotic diarrhea |
|
Vitamin D |
Serum calcium > 11 mg/dl |
|
Fluoride |
Children: moderate dental fluorosis Adults: moderate skeletal fluorosis |
|
Niacin |
Flushing |
|
Vitamin B6 |
Sensory neuropathy |
|
Synthetic Folic Acid |
Neuropathy in B12-deficient individuals |
|
Choline |
Hypotension, fishy body odor |
When data were not available on children or adolescents, ULs were determined by extrapolating from the UL for adults based on body weight differences using the formula
ULchild = (ULadult)(Weightchild/Weightadult).
The reference weight for males aged 19 through 30 years (see Appendix III) was used for adults and the reference weights for female children and adolescents were used in the formula above to obtain the UL for each age group. The use of these reference weights yields a conservative UL to protect the sensitive individuals in each age group.
REFERENCES
Barger-Lux MJ, Heaney RP, Lanspa SJ, Healy JC, DeLuca HF. 1995. An investigation of sources of variation in calcium absorption efficiency. J Clin Endocrinol Metab 80:406-411.
Dourson ML, Stara JF. 1983. Regulatory history and experimental support of uncertainty (safety) factors. Regul Toxicol Pharmacol 3:224–238.
EPA (U.S. Environmental Protection Agency). 1996. Proposed Guidelines for Carcinogen Risk Assessment; Notice. Federal Register 61:17960–18011.
FAO/WHO (Food and Agriculture Organization of the United Nations/World Health Organization). 1982. Evaluation of certain food additives and contaminants. Twenty-sixth report of the Joint FAO/WHO Expert Committee on Food Additives (WHO Technical Report Series No. 683).
FAO/WHO (Food and Agriculture Organization of the United Nations/World Health Organization). 1995. The application of risk analysis to food standard issues. Recommendations to the Codex Alimentarius Commission (ALINORM 95/9, Appendix 5). Geneva: World Health Organization.
Health Canada. 1993. Health Risk Determination—The Challenge of Health Protection. Ottawa: Health Canada, Health Protection Branch.
Hill AB. 1971. Principles of Medical Statistics, 9th ed. New York: Oxford University Press.
IOM (Institute of Medicine). 1994. How Should the Recommended Dietary Allowances Be Revised? Food and Nutrition Board. Washington, DC: National Academy Press.
IOM (Institute of Medicine). 1997. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, DC: National Academy Press.
IOM (Institute of Medicine). 1998. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academy Press.
Klaassen CD, Amdur MO, Doull J. 1986. Casarett and Doull’s Toxicology: The Basic Science of Poisons, 3d ed. New York: Macmillan.
Mertz W, Abernathy CO, Olin SS. 1994. Risk Assessment of Essential Elements. Washington, DC: ILSI Press.
Narang NK, Gupta RC, Jain MK. 1984. Role of vitamin D in pulmonary tuberculosis. J Assoc Physicians India 32:185-188.
NRC (National Research Council). 1983. Risk Assessment in the Federal Government: Managing the Process. Washington, DC: National Academy of Sciences.
NRC (National Research Council). 1989. Diet and Health: Implications for Reducing Chronic Disease Risk. Report of the Committee on Diet and Health, Food and Nutrition Board, Commission on Life Sciences. Washington, DC: National Academy Press.
NRC (National Research Council). 1994. Science and Judgment in Risk Assessment. Committee on Risk Assessment of Hazardous Air Pollutants. Board on Environmental Studies and Toxicology. Washington, DC: National Academy of Sciences.
OTA (Office of Technology Assessment). 1993. Researching Health Risks. Washington, DC: Office of Technology Assessment.
WHO (World Health Organization). 1987. Principles for the Safety Assessment of Food Additives and Contaminants in Food. Environmental Health Criteria 70. Geneva: World Health Organization.
WHO (World Health Organization). 1996. Trace Elements in Human Nutrition and Health. Geneva. Prepared in collaboration with the Food and Agriculture Organization of the United Nations and the International Atomic Energy Agency. Geneva: World Health Organization.
Zielhuis RL, van der Kreek FW. 1979. The use of a safety factor in setting health-based permissible levels for occupational exposure. Int Arch Occup Environ Health 42:191–201.
Appendix I Recommended Dietary Intakes For Individuals![]()
Recommended Dietary Intakes for Individuals
|
Life-Stage |
Calcium |
Phosphorus |
Magnesium |
Vitamin D |
Fluoride |
Thiamin |
Riboflavin |
Niacin |
B6 Vitamin B6 |
Folate |
Vitamin B12 |
Pantothenic |
Biotin |
Choline e |
|||||
|
Group |
(mg/d) |
(mg/d) |
(mg/d) |
(m g/d) a,b |
(mg/d) |
(mg/d) |
(mg/d) |
(mg/d) c |
(mg/d) |
(m g/d) d |
(m g/d) |
Acid (mg/d) |
(m g/d) |
(mg/d) |
|||||
|
Infants 0–6 mo |
210* |
100* |
30* |
5* |
0.01* |
0.2* |
0.3* |
2* |
0.1* |
65* |
0.4* |
1.7* |
5* |
125* |
|||||
|
7–12 mo |
270* |
275* |
75* |
5* |
0.5* |
0.3* |
0.4* |
4* |
0.3* |
80* |
0.5* |
1.8* |
6* |
150* |
|||||
|
Children 1–3 yr |
500* |
460 |
80 |
5* |
0.7* |
0.5 |
0.5 |
6 |
0.5 |
150 |
0.9 |
2* |
8* |
200* |
|||||
|
4–8 yr |
800* |
500 |
130 |
5* |
1* |
0.6 |
0.6 |
8 |
0.6 |
200 |
1.2 |
3* |
12* |
250* |
|||||
|
Males 9–13 yr |
1,300* |
1,250 |
240 |
5* |
2* |
0.9 |
0.9 |
12 |
1.0 |
300 |
1.8 |
4* |
20* |
375* |
|||||
|
14–18 yr |
1,300* |
1,250 |
410 |
5* |
3* |
1.2 |
1.3 |
16 |
1.3 |
400 |
2.4 |
5* |
25* |
550* |
|||||
|
19–30 yr |
1,000* |
700 |
400 |
5* |
4* |
1.2 |
1.3 |
16 |
1.3 |
400 |
2.4 |
5* |
30* |
550* |
|||||
|
31–50 yr |
1,000* |
700 |
420 |
5* |
4* |
1.2 |
1.3 |
16 |
1.3 |
400 |
2.4 |
5* |
30* |
550* |
|||||
|
51–70 yr |
1,200* |
700 |
420 |
10* |
4* |
1.2 |
1.3 |
16 |
1.7 |
400 |
2.4f |
5* |
30* |
550* |
|||||
|
> 70 yr |
1,200* |
700 |
420 |
15* |
4* |
1.2 |
1.3 |
16 |
1.7 |
400 |
2.4f |
5* |
30* |
550* |
|||||
|
Females 9–13 yr |
1,300* |
1,250 |
240 |
5* |
2* |
0.9 |
0.9 |
12 |
1.0 |
300 |
1.8 |
4* |
20* |
375* |
|||||
|
14–18 yr |
1,300* |
1,250 |
360 |
5* |
3* |
1.0 |
1.0 |
14 |
1.2 |
400g |
2.4 |
5* |
25* |
400* |
|||||
|
19–30 yr |
1,000* |
700 |
310 |
5* |
3* |
1.1 |
1.1 |
14 |
1.3 |
400g |
2.4 |
5* |
30* |
425* |
|||||
|
31–50 yr |
1,000* |
700 |
320 |
5* |
3* |
1.1 |
1.1 |
14 |
1.3 |
400g |
2.4 |
5* |
30* |
425* |
|||||
|
51–70 yr |
1,200* |
700 |
320 |
10* |
3* |
1.1 |
1.1 |
14 |
1.5 |
400 |
2.4f |
5* |
30* |
425* |
|||||
|
> 70 yr |
1,200* |
700 |
320 |
15* |
3* |
1.1 |
1.1 |
14 |
1.5 |
400 |
2.4f |
5* |
30* |
425* |
|||||
|
Pregnancy |
|||||||||||||||||||
|
£ 18 yr |
1,300* |
1,250 |
400 |
5* |
3* |
1.4 |
1.4 |
18 |
1.9 |
600h |
2.6 |
6* |
30* |
450* |
|||||
|
19–30 yr |
1,000* |
700 |
350 |
5* |
3* |
1.4 |
1.4 |
18 |
1.9 |
600h |
2.6 |
6* |
30* |
450* |
|||||
|
31–50 yr |
1,000* |
700 |
360 |
5* |
3* |
1.4 |
1.4 |
18 |
1.9 |
600h |
2.6 |
6* |
30* |
450* |
|||||
|
Lactation |
|||||||||||||||||||
|
£ 18 yr |
1,300* |
1,250 |
360 |
5* |
3* |
1.5 |
1.6 |
17 |
2.0 |
500 |
2.8 |
7* |
35* |
550* |
|||||
|
19–30 yr |
1,000* |
700 |
310 |
5* |
3* |
1.5 |
1.6 |
17 |
2.0 |
500 |
2.8 |
7* |
35* |
550* |
|||||
|
31–50 yr |
1,000* |
700 |
320 |
5* |
3* |
1.5 |
1.6 |
17 |
2.0 |
500 |
2.8 |
7* |
35* |
550* |
|||||
|
|
|
||||||||||||||||||
NOTE: This table presents Recommended Dietary Allowances (RDAs) in bold type and Adequate Intakes (AIs) in ordinary type followed by an asterisk (*). RDAs and AIs may both be used as goals for individual intake. RDAs are set to meet the needs of almost all (97 to 98 percent) individuals in a group. For healthy breastfed infants, the AI is the mean intake. The AI for other life-stage and gender groups is believed to cover needs of all individuals in the group, but lack of data or uncertainty in the data prevent being able to specify with confidence the percentage of individuals covered by this intake. a
b
In the absence of adequate exposure to sunlight.c
As niacin equivalents (NE). 1 mg of niacin = 60 mg of tryptophan; 0-6 months = preformed niacin (not NE).d
As dietary folate equivalents (DFE). 1 DFE = 1 m g food folate = 0.6 m g of folic acid (from fortified food or supplement) consumed with food = 0.5 m g of synthetic (supplemental) folic acid taken on an empty stomach.e
Although AIs have been set for choline, there are few data to assess whether a dietary supply of choline is needed at all stages of the life cycle, and it may be that the choline requirement can be met by endogenous synthesis at some of these stages.f
Because 10 to 30 percent of older people may malabsorb food-bound B12, it is advisable for those older than 50 years to meet their RDA mainly by consuming foods fortified with B12 or a supplement containing B12.g
In view of evidence linking folate intake with neural tube defects in the fetus, it is recommended that all women capable of becoming pregnant consume 400 m g of synthetic folic acid from fortified foods and/or supplements in addition to intake of food folate from a varied diet.h
It is assumed that women will continue consuming 400 m g of folic acid until their pregnancy is confirmed and they enter prenatal care, which ordinarily occurs after the end of the periconceptional period—the critical time for formation of the neural tube.Appendix II
![]()
Options for Dealing with Uncertainties
Methods for dealing with uncertainties in scientific data are generally understood by working scientists and require no special discussion here, except to point out that such uncertainties should be explicitly acknowledged and taken into account whenever a risk assessment is undertaken. More subtle and difficult problems are created by uncertainties associated with some of the inferences that need to be made in the absence of directly applicable data; much confusion and inconsistency can result if they are not recognized and dealt with in advance of undertaking a risk assessment.
The most significant inference uncertainties arise in risk assessments whenever attempts are made to answer the following questions (NRC, 1994):
Depending on the nutrient under review, at least partial, empirically based answers to some of these questions may be available, but in no case is scientific information likely to be sufficient to provide a highly certain answer; in many cases there will be no relevant data for the nutrient in question.
It should be recognized that, for several of these questions, certain inferences have been widespread for long periods of time, and thus, it may seem unnecessary to raise these uncertainties anew. When several sets of animal toxicology data are available, for example, and data are insufficient to identify the set (i.e., species, strain, adverse effects endpoint) that "best" predicts human response, it has become traditional to select that set in which toxic responses occur at lowest dose ("most sensitive"). In the absence of definitive empirical data applicable to a specific case, it is generally assumed that there will not be more than a 10-fold variation in response among members of the human population. In the absence of absorption data, it is generally assumed that humans will absorb the chemical at the same rate as the animal species used to model human risk. In the absence of complete understanding of biological mechanisms, it is generally assumed that, except possibly for certain carcinogens, a threshold dose must be exceeded before toxicity is expressed. These types of long-standing assumptions, which are necessary to complete a risk assessment, are recognized by risk assessors as attempts to deal with uncertainties in knowledge (NRC, 1994).
A past National Research Council (NRC) report (1983) recommended the adoption of the concepts and definitions that have been discussed in this report. The NRC committee recognized that throughout a risk assessment, data and basic knowledge will be lacking and that risk assessors will be faced with several scientifically plausible options (called "inference options" by the NRC) for dealing with questions such as those presented above. For example, several scientifically supportable options for dose-scaling across species and for high-to-low dose extrapolation, but no ready means to identify those that are clearly best supported. The NRC committee recommended that regulatory agencies in the United States identify the needed inference options in risk-assessment and specify, through written risk assessment guidelines, the specific options that will be used for all assessments. Agencies in the United States have identified the specific models to be used to fill gaps in data and knowledge; these have come to be called default options (EPA, 1986).
The use of defaults to fill knowledge and data gaps in risk assessment has the advantage of ensuring consistency in approach (the same defaults are used for each assessment) and for minimizing or eliminating case-by-case manipulations of the conduct of risk assessment to meet predetermined risk management objectives. The major disadvantage of the use of defaults is the potential for displacement of scientific judgment by excessively rigid guidelines. A remedy for this disadvantage was also suggested by the NRC committee: risk assessors should be allowed to replace defaults with alternative factors in specific cases of chemicals for which relevant scientific data were available to support alternatives. The risk assessors’ obligation in such cases is to provide explicit justification for any such departure. Guidelines for risk assessment issued by the U.S. Environmental Protection Agency, for example, specifically allow for such departures (EPA, 1986).
The use of preselected defaults is not the only way to deal with model uncertainties. Another option is to allow risk assessors complete freedom to pursue whatever approaches they judge applicable in specific cases. Because many of the uncertainties cannot be resolved scientifically, case-by-case judgments without some guidance on how to deal with them will lead to difficulties in achieving scientific consensus, and the results of the assessment may not be credible.
Another option for dealing with uncertainties is to allow risk assessors to develop a range of estimates, based on application of both defaults and alternative inferences that, in specific cases, have some degree of scientific support. Indeed, appropriate analysis of uncertainties would seem to require such a presentation of risk results. Although presenting a number of plausible risk estimates has clear advantages in that it would seem to reflect more faithfully the true state of scientific understanding, there are no well-established criteria for using such complex results in risk management.
The various approaches to dealing with uncertainties inherent to risk assessment, and discussed in the foregoing sections, are summarized in Table II-1.
Table II–1. Approaches for Dealing with Uncertainties in a Risk-Assessment Program
|
Program Model |
Advantages |
Disadvantages |
|
Case-by-case judgments by experts |
Flexibility High potential to maximize use of most relevant scientific information bearing on specific issues |
Potential for inconsistent treatment of different issues Difficulty in achieving consensus Need to agree on defaults
|
|
Written guidelines specifying defaults for data and model uncertainties (with allowance for departures in specific cases) |
Consistent treatment of different issues Maximize transparency of process Allow resolution of scientific dis-agreements by resort to defaults |
May be difficult to justify departure, or to achieve consensus among scientists that departures are justified in specific cases Danger that uncertainties will be overlooked |
|
Assessors asked to present full array of estimates, using all scientifically plausible models |
Maximize use of scientific infor-mation Reasonably reliable portrayal of true state of scientific understanding |
Highly complex characterization of risk, with no easy way to discrimin-ate among estimates Size of required effort may not be commensurate with utility of the outcome |
Specific default assumptions for assessing nutrient risks have not been recommended. Rather, the approach calls for case-by-case judgments, with the recommendation that the basis for the choices made be explicitly stated. Some general guidelines for making these choices will, however, be offered.
References
EPA (U.S. Environmental Protection Agency). 1986. Guidelines for Carcinogen Risk Assessment. Federal Register 51:33992–34003.
NRC (National Research Council). 1983. Risk Assessment in the Federal Government: Managing the Process. Washington, DC: National Academy of Sciences.
NRC (National Research Council). 1994. Science and Judgment in Risk Assessment. Committee on Risk Assessment of Hazardous Air Pollutants. Board on Environmental Studies and Toxicology. Washington, DC: National Academy of Sciences.
Appendix III
![]()
Reference Heights and Weights for
Children and Adults
|
Gender |
Age |
Median Body Mass Index, kg/m2 |
Reference Height, cm (in) |
ReferenceWeight,a. kg (lb) |
|
Male, female |
2–6 mo |
– |
64 (25) |
7 (16) |
|
7–11 mo |
– |
72 (28) |
9 (20) |
|
|
1–3 yr |
– |
91 (36) |
13 (29) |
|
|
4–8 yr |
15.8 |
118 (46) |
22 (48) |
|
|
Male |
9–13 yr |
18.5 |
147 (58) |
40 (88) |
|
14–18 yr |
21.3 |
174 (68) |
64 (142) |
|
|
19–30 yr |
24.4 |
176 (69) |
76 (166) |
|
|
Female |
9–13 yr |
18.3 |
148 (58) |
40 (88) |
|
14–18 yr |
21.3 |
163 (64) |
57 (125) |
|
|
19–30 yr |
22.8 |
163 (64) |
61 (133) |
Adapted from: NHANES III, 1988-1994, unpublished data, R. Briefel, National Center for Health Statistics, Centers for Disease Control and Prevention, 1997.
a
Calculated from median body mass index and median heights for ages 4–8 yr and older.
Appendix IV
![]()
Case Studies of Application of Model of
Risk Assessment for Nutrients
1
A. CALCIUM
Hazard Identification
Calcium is among the most ubiquitous of elements found in the human system. Calcium plays a major role in the metabolism of virtually every cell in the body and interacts with a large number of other nutrients. As a result, disturbances of calcium metabolism give rise to a wide variety of adverse reactions. Disturbances of calcium metabolism, particularly those that are characterized by changes in extracellular ionized calcium concentration, can cause damage in the function and structure of many organs and systems.
Currently, the available data on the adverse effects of excess calcium intake in humans primarily concerns calcium intake from nutrient supplements. Of the many possible adverse effects of excessive calcium intake, the three most widely studied and biologically important are: kidney stone formation (nephrolithiasis), the syndrome of hypercalcemia and renal insufficiency with and without alkalosis (referred to historically as milk-alkali syndrome when associated with a constellation of peptic ulcer treatments), and the interaction of calcium with the absorption of other essential minerals. These are not the only adverse effects associated with excess calcium intake. However, the vast majority of reported effects are related to or result from one of these three conditions.
Nephrolithiasis
Twelve percent of the U.S. population will form a renal stone over their lifetime (Johnson et al., 1979), and it has generally been assumed that nephrolithiasis is, to a large extent, a nutritional disease. Research over the last 40 years has shown that there is a direct relationship between periods of affluence and increased nephrolithiasis (Robertson, 1985). A number of dietary factors seem to play a role in determining the incidence of this disease. In addition to being associated with increased calcium intakes, nephrolithiasis appears to be associated with higher intakes of oxalate, protein, and vegetable fiber (Massey et al., 1993). Goldfarb (1994) argued that dietary calcium plays a minor role in nephrolithiasis because only 6 percent of the overall calcium load appears in the urine of normal individuals. Also, the efficiency of calcium absorption is substantially lower when calcium supplements are consumed (Sakhaee et al., 1994).
The issue is made more complex by the association between high sodium intakes and hypercalciuria, since sodium and calcium compete for reabsorption at the same sites in the renal tubules (Goldfarb, 1994). Other minerals, such as phosphorus and magnesium, also are risk factors in stone formation (Pak, 1988). These findings suggest that excess calcium intake may play only a contributing role in the development of nephrolithiasis.
Two recent companion prospective epidemiologic studies in men (Curhan et al., 1993) and women (Curhan et al., 1997) with no history of kidney stones found that intakes of dietary calcium greater than 1,050 mg (26.3 mmol)/day in men and greater than 1,098 mg (27.5 mmol)/day in women were associated with a reduced risk of symptomatic kidney stones. This association for dietary calcium was attenuated when the intake of magnesium and phosphorus were included in the model for women (Curhan et al., 1997). This apparent protective effect of dietary calcium is attributed to the binding by calcium in the intestinal lumen of oxalate, which is a critical component of most kidney stones. In contrast, Curhan et al. (1997) found that after adjustment for age, intake of supplemental calcium was associated with an increased risk for kidney stones. After adjustment for potential confounders, the relative risk among women who took supplemental calcium, compared with women who did not, was 1.2. Calcium supplements may be taken without food, which limits opportunity for the beneficial effect of binding oxalate in the intestine. A similar effect of supplemental calcium was observed in men (Curhan et al., 1993) but failed to reach statistical significance. Neither study controlled for the time that calcium supplements were taken (for example, with or without meals); thus, it is possible that the observed significance of the results in women may be due to different uses of calcium supplements by men and women. Clearly, more carefully controlled studies are needed to determine the strength of the causal association between calcium intake vis-à-vis the intake of other nutrients and kidney stones in healthy individuals.
The association between calcium intake and urinary calcium excretion is weaker in children than in adults. However, as observed in adults, increased levels of dietary sodium are significantly associated with increased urinary calcium excretion in children (Matkovic et al., 1995, O’Brien et al., 1996).
Hypercalcemia and Renal Insufficiency (Milk-Alkali Syndrome)
The syndrome of hypercalcemia and, consequently, renal insufficiency with or without metabolic alkalosis is associated with severe clinical and metabolic derangements affecting virtually every organ system (Orwoll, 1982). Renal failure may be reversible but may also be progressive if the syndrome is unrelieved. Progressive renal failure may result in the deposition of calcium in soft tissues including the kidney (for example, nephrocalcinosis) with a potentially fatal outcome (Junor and Catto, 1976). This syndrome was first termed milk-alkali syndrome (MAS) in the context of the high milk and absorbable antacid intake which derived from the "Sippy diet" regimen for the treatment of peptic ulcer disease. MAS needs to be distinguished from primary hyperparathyroidism, in which primary abnormality of the parathyroid gland results in hypercalcemia, metabolic derangement, and impaired renal calcium resorption. As the treatment of peptic ulcers has changed (for example, systemically absorbed antacids and large quantities of milk are now rarely prescribed), the incidence of this syndrome has decreased (Whiting and Wood, 1997).
A review of the literature revealed 26 reported cases of MAS linked to high calcium intake from supplements and food since 1980 without other causes of underlying renal disease (Table IV-1). These reports described what appears to be the same syndrome at supplemental calcium intakes of 1.5 to 16.5 g (37.5 to 412.5 mmol)/day for 2 days to 30 years. Estimates of the occurrence of MAS in the North American population may be low since mild cases are often overlooked and the disorder may be confused with a number of other syndromes presenting with hypercalcemia.
No reported cases of MAS in children were found in the literature. This was not unexpected since children have very high rates of bone turnover and calcium utilization relative to adults (Abrams et al., 1992). A single case of severe constipation directly linked to daily calcium supplementation of 1,000 mg (25 mmol) or more has been reported in an 8-year-old boy, but this may represent an idiosyncratic reaction of calcium ions exerted locally in the intestine or colon (Frithz et al., 1991).
Calcium/Mineral Interactions
Calcium interacts with iron, zinc, magnesium, and phosphorus (Clarkson et al., 1967; Hallberg et al., 1992; Schiller et al., 1989; Spencer et al., 1965). Calcium-mineral interactions are more difficult to quantify than nephrolithiasis and MAS, since in many cases the interaction of calcium with several other nutrients results in changes in the absorption and utilization of each. Thus, it is virtually impossible to determine a dietary level at which calcium intake alone disturbs the absorption or metabolism of other minerals. Nevertheless, calcium clearly inhibits iron absorption in a dose-dependent and dose-saturable fashion (Hallberg et al., 1992). However, the available human data fail to show cases of iron deficiency or even reduced iron stores as a result of calcium intake (Snedeker et al., 1982; Sokoll and Dawson-Hughes, 1992). Similarly, except for a single report of negative zinc balance in the presence of calcium supplementation (Wood and Zheng, 1990), the effects of calcium on zinc absorption have not been shown to be associated with zinc depletion or undernutrition. Neither have interactions of high levels of calcium with magnesium or phosphorus shown evidence of depletion of the affected nutrient (Shils, 1994).
Thus, in the absence of clinically or functionally significant depletion of the affected nutrient, calcium interaction with other minerals represents a potential risk rather than an adverse effect, in the sense that nephrolithiasis or hypercalcemia are adverse effects. Still, the potential for increased risk of mineral depletion in vulnerable populations such as those on
Table IV-1. Case Reports of Milk Alkali Syndrome (single dose reported)a
|
Studies |
Ca Intake (g/day)b |
Duration |
Mitigating Factors |
Brandwein and Sigman, 1994 |
2.7c |
2 years, 8 months |
none reported |
Campbell et al., 1994 |
5d |
3 months |
none reported |
French et al., 1986 |
8c |
2 years |
none reported |
|
4.2c |
>2 years |
thiazide |
|
Kallmeyer and Funston, 1983 |
8d |
10 years |
alkali in antacid |
Kapsner et al., 1986 |
10d |
10 months |
none reported |
|
6.8d |
7 months |
none reported |
|
|
4.8c |
2 days |
10 year history of antacid use |
|
Muldowney and Mazbar, 1996 |
1.7c |
13 months (52 weeks) |
none reported |
Whiting and Wood, 1997 |
2.4c |
> 1 year |
none reported |
Schuman and Jones, 1985 |
9.8d |
20 years |
none reported |
|
4.8d |
6 weeks |
10 year history of antacid intake |
|
Abreo et al., 1993 |
9.6c |
> 3 months |
none reported |
|
3.6c |
> 2 years |
none reported |
|
|
10.8d |
Not stated |
none reported |
|
Carroll et al., 1983 |
4.2d |
30 years |
none reported |
|
2c |
5 years |
none reported |
|
|
3.8d |
2 months |
vitamins A and E |
|
|
2.8d |
10 years |
NaHCO3, 5 g/d |
|
Whiting and Wood, 1997 |
2.3–4.6c |
> 1 year |
none reported |
Lin et al., 1996 |
1.5c |
4 weeks |
none reported |
Kleinman et al., 1991 |
16.5d |
2 weeks |
10 year history of antacid use |
Hart et al., 1982 |
10.6d |
Not stated |
NaHCO3, 2 g/d |
Bullimore and Miloszewski, 1987 |
6.5d |
23 years |
alkali in antacid |
Gora et al., 1989 |
4c |
2 years |
thiazide |
Number of Subjects |
26 |
– |
– |
Mean (SD) |
5.9 |
3 years, 8 months |
– |
Median |
4.8 |
13 months |
– |
Range |
1.5–>16.5 |
2 days–23 years |
– |
a
b
Intake estimates provided by Whiting and Wood (1997).c
Calcium intake from supplements reported only.d
Calcium intake from supplements and diet reported (for example, milk and yogurt consumption). Other dietary sources of calcium not reported are not included.
very low mineral intakes or the elderly needs to be incorporated into the uncertainty factor in deriving a UL for calcium. Furthermore, because of their potential to increase the risk of mineral depletion in vulnerable populations, calcium-mineral interactions should be the subject of additional studies.
Dose-Response Assessment
Adults: Ages 19 through 70 Years
Data Selection. Based on the discussion of adverse effects of excess calcium intake above, the most appropriate data available for identifying a critical endpoint and a NOAEL (or LOAEL) concern risk of MAS or nephrolithiasis. There are few well-controlled, chronic studies of calcium that show a dose-response relationship. While there are inadequate data on nephrolithiasis to establish a dose-response relationship and to identify a NOAEL (or LOAEL), there are adequate data on MAS that can be used.
Identification of a NOAEL (or LOAEL) and Critical Endpoint. Using MAS as the clinically defined critical endpoint, a LOAEL in the range of 4 to 5 grams (100 to 125 mmol)/day can be identified for adults (Table IV-2). A review of these reports revealed calcium intakes from supplements (and in some cases from dietary sources as well) in the range of 1.5 to 16.5 g (37.5 to 412.5 mmol)/day. A median intake of 4.8 g (120 mmol)/day resulted in documented cases. Since many of these reports included dietary calcium intake as well as intake from supplements, an intake in the range of 5 g (125 mmol)/day represents a LOAEL for total calcium intake (for example, from both supplements and food). A solid figure for a NOAEL is not available, but researchers have observed that daily calcium intakes of 1,500 to 2,400 mg (37.5 to 60 mmol) (including supplements), used to treat or prevent osteoporosis, did not result in hypercalcemic syndromes (Kochersberger et al., 1991; McCarron and Morris, 1985; Riggs et al., 1996; Saunders et al., 1988; Smith et al., 1989; Thys-Jacobs et al., 1989).
TABLE IV-2. Case Reports of Milk Alkali Syndrome (Multi- and increasing doses reported)
|
Ca Intake (Dose 1) (g/day) |
Duration (months) |
Ca Intake (Dose 2) (g/day) |
Duration |
|
Beall and Scofield, 1995 |
1a |
13 |
2.4a |
2 weeks |
|
1 |
13 |
4.2 |
2 weeks |
|
|
0.3a |
6 |
1.8a |
1 month |
|
Hakim et al., 1979 |
1a |
13 |
2.5a |
3.5 weeks |
Newmark and Nugent, 1993 |
not reported |
13 |
8.4a |
< 1 year (recent) |
Schuman and Jones, 1985 |
not reported |
13 |
4.6 |
6 weeks |
Carroll et al., 1983 |
2.5 |
13 |
3 |
13 months |
Malone and Horn, 1971 |
not reported |
13 |
3a |
4.5 weeks |
|
Dorsch, 1986 |
not reported |
13 |
2.1a |
6 months |
|
Number of Subjects |
9 |
9 |
||
|
Mean (SD) |
1.2 (0.8) |
12 |
3.6(2.0) |
16.7 (21) |
|
Median |
1 |
13 |
3 |
4.5 |
|
Range |
0.3–2.5 |
6–13 |
1.8–8.4 |
2–53 |
a
Consideration of hypercalciuria may have additional relevance to the derivation of a UL for adults. Hypercalciuria is observed in approximately 50 percent of patients with calcium oxalate/apatite nephrolithiasis and is an important risk factor for nephrolithiasis (Lemann et al., 1991; Whiting and Wood, 1997). Therefore, it is plausible that high calcium intakes associated with hypercalciuria could produce nephrolithiasis. Burtis et al. (1994) reported a significant positive association between both dietary calcium and sodium intake and hypercalciuria in 282 renal stone patients and derived a regression equation to predict the separate effects of dietary calcium and urinary sodium on urinary calcium excretion. Setting urinary sodium excretion at 150 mmol/day and defining hypercalciuria for men as greater than 300 mg (7.5 mmol) of calcium per day excreted (Burtis et al., 1994), the calcium intake that would be associated with hypercalciuria was 1,685 mg (42.1 mmol)/day. For women, for whom hypercalcemia was defined as greater than 250 mg (6.2 mmol)/day excreted, it would be 866 mg (21.6 mmol)/day. The results of these calculations from the Burtis et al. (1994) equation suggest that calcium intakes lower than adequate intake levels derived earlier in this chapter for females could result in hypercalciuria in susceptible individuals.
Although Burtis et al. (1994) identified what could be defined as LOAELs for hypercalciuria, 1,685 mg (42.1 mmol)/day in men and 866 mg (21.6 mmol)/day in women, these values are not considered as appropriate for use as the LOAEL for healthy adults as they were based on patients with renal stones. However, they support for the need for conservative estimates of the Tolerable Upper Intake Level (UL).
Uncertainty Assessment. An uncertainty factor (UF) of 2 is recommended to take into account the potential for increased risk of high calcium intake in these areas: (1) the 12 percent of the American population with renal stones, (2) the occurrence of hypercalciuria with intakes as low as 1,700 mg (42.5 mmol)/day in male and 870 mg (21.7 mmol)/day in female patients with renal stones (Burtis et al., 1994), and (3) the potential to increase the risk of mineral depletion in vulnerable populations due to the interference of calcium on mineral bioavailability, especially iron and zinc.
Derivation of the UL. A UL of 2.5 g (62.5 mmol)/day is calculated by dividing a LOAEL of 5 g (125 mmol)/day by the UF of 2. The data summarized in Table IV-2 show that calcium intakes of 0.3 to 2.5 g (7.5 to 62.5 mmol)/day will not cause MAS and provide supportive evidence for a UL of 2,500 mg (62.5 mmol)/day for adults. The estimated UL for calcium in adults is judged to be conservative. For individuals who are particularly susceptible to high calcium intakes, such as those with hypercalcemia and hyperabsorptive hypercalciuria, this level or below should be protective.
UL for Adults Ages 19–70 years 2,500 mg (62.5 mmol)/day
Infants: Ages 0 to 12 Months
The safety of calcium intakes above the levels provided by infant formulas and weaning foods has recently been studied by Dalton et al. (1997). They did not find any effect on iron status from calcium intakes of approximately 1,700 mg (42.5 mmol)/day in infants, which was attained using calcium-fortified infant formula. However, further studies are needed before a UL specific to infants can be established.
UL for Infants Ages 0–12 months Not possible to establish for supplementary calcium
Toddlers, Children, and Adolescents: Ages 1 through 18 years
Although the safety of excess calcium intake in children aged 1 through 18 years has not been studied, a UL of 2,500 mg (62.5 mmol)/day is recommended for these life-stage groups. Although calcium supplementation in children may appear to pose minimal risk of MAS or hyperabsorptive hypercalciuria, risk of depletion of other minerals associated with high calcium intakes, may be greater. With high calcium intake, small children may be especially susceptible to deficiency of iron and zinc (Golden and Golden, 1981; Schlesinger et al., 1992; Simmer et al., 1988). However, no dose-response data exist regarding these interactions in children or the development of adaptation to chronic high calcium intakes. After age 9, rates of calcium absorption and bone formation begin to increase in preparation for pubertal development, but a conservative UL of 2,500 mg (62.5 mmol)/day (from diet and supplements) is recommended for children due to the lack of data.
UL for Children Ages 1–18 years 2,500 mg (62.5 mmol)/day
Older Adults: Ages > 70 Years
Several physiologic differences in older adults need to be considered in setting the UL for people over age 70. Because this population is more likely to have achlorhydria (Recker, 1985), absorption of calcium, except when associated with meals, is likely to be somewhat impaired, which would protect these individuals from the adverse effects of high calcium intakes. Furthermore, there is a decline in calcium absorption associated with age that results from changes in function of the intestine (Ebeling et al., 1994). However, the elderly population is also more likely to have marginal zinc status, which theoretically would make them more susceptible to the negative interactions of calcium and zinc (Wood and Zheng, 1990). This matter deserves more study. These effects serve to increase the UF on the one hand and decrease it on the other, with the final result being to use the same UL for older adults as for younger adults.
UL for Older Adults Ages > 70 years 2,500 mg (62.5 mmol)/day
Pregnancy and Lactation
The available data were judged to be inadequate for deriving a UL for pregnant and lactating women that is different from the UL for the nonpregnant and nonlactating female.
UL for Pregnancy Ages 14–50 years 2,500 mg (62.5 mmol)/day
UL for Lactation Ages 14–50years 2,500 mg (62.5 mmol)/day
Special Considerations
Not surprisingly, the ubiquitous nature of calcium results in a population of individuals with a wide range of sensitivities to its toxic effects. Subpopulations known to be particularly susceptible to the toxic effects of calcium include individuals with renal failure, those using thiazide diuretics (Whiting and Wood, 1997), and those with low intakes of minerals that interact with calcium (for example, iron, magnesium, zinc). For the majority of the general population, intakes of calcium from food substantially above the UL are probably safe.
Exposure Assessment
The highest median intake of calcium for any age group found in the 1994 CSFII data, adjusted for day-to-day variation (Nusser et al., 1996), was for boys 14 through 18 years of age with a median intake of 1,094 mg (27.4 mmol)/day and a ninety-fifth percentile intake of 2,039 mg (51 mmol)/day. Calcium supplements were used by less than 8 percent of young children, 14 percent of men, and 25 percent of women in the United States (Moss et al., 1989). Daily dosages from supplements at the ninety-fifth percentile were relatively small for children (160 mg [4 mmol]), larger for men (624 mg /[15.6 mmol]), and largest for women (904 mg [22.6 mmol]) according to Moss et al. (1989).
Risk Characterization
Although the ninety-fifth percentile of daily intake did not exceed the UL for any age group (2,101 mg [52.5 mmol] in males 14 through 18 years old) in the 1994 CSFII data, persons with a very high caloric intake, especially if intakes of dairy products were also high, may exceed the UL of 2,500 mg (62.5 mmol)/day.
Even if the ninety-fifth percentile of intake from foods and the most recently available estimate of the ninety-fifth percentile of supplement use (Moss et al., 1989) are added together for teenage boys (1,920 + 928 mg/day) or for teenage girls (1,236 + 1,200 mg/day), total intakes are just at or slightly above the UL. Although users of dietary supplements (of any kind) tend to also have higher intakes of calcium from food than nonusers (Slesinski et al., 1996), it is unlikely that the same person would fall at the upper end of both ranges. Furthermore, the prevalence of usual intakes (from foods plus supplements) above the UL is well below 5 percent, even for age groups with relatively high intakes. Nevertheless, an informal survey of food products in supermarkets in the Washington, D.C. metropolitan area between 1994 and 1996 showed that the number of calcium-fortified products doubled in the two year period (Y. Park, Food and Drug Administration, February 1997, personal communication). Therefore, it is important to maintain surveillance of the calcium-fortified products in the marketplace and monitor their impact on calcium intake.
References
Abrams SA, Esteban NV, Vieira NE, Sidbury JB, Specker BL, Yergey AL. 1992. Developmental changes in calcium kinetics in children assessed using stable isotopes. J Bone Miner Res 7:287–293.
Abreo K, Adlakha A, Kilpatrick S, Flanagan R, Webb R, Shakamuri S. 1993. The Milk-Alkali Syndrome. A reversible form of acute renal failure. Arch Intern Med 153:1005–1010.
Beall DP, Scofield RH. 1995. Milk-alkali syndrome associated with calcium carbonate consumption: Report of 7 patients with parathyroid hormone levels and an estimate of prevalence among patients hospitalized with hypercalcemia. Medicine 74:89–96.
Brandwein SL, Sigman, KM. 1994. Case report: Milk-alkali syndrome and pancreatitis. Am J Med Sci 308:173–176.
Bullimore DW, Miloszewski KJ. 1987. Raised parathyroid hormone levels in the milk-alkali syndrome: An appropriate response? Postgrad Med J 63:789–792.
Burtis WJ, Gay L, Insogna KL, Ellison A, Broadus AE. 1994. Dietary hypercalciuria in patients with calcium oxalate kidney stones. Am J Clin Nutr 60:424–429.
Campbell SB, MacFarlane DJ, Fleming SJ, Khafagi FA. 1994. Increased skeletal uptake of Tc-99m Methylene Disphosphonate in Milk-Alkali Syndrome. Clin Nucl Med 19:207–211.
Carroll MD, Abraham S, Dresser CM. 1983. Dietary intake source data: United States, 1976–1980. Data from the National Health Survey. Vital and Health Statistics series 11, no. 231. DHHS Publ. No. (PHS) 83-1681. Hyattsville, MD: National Center for Health Statistics, Public Health Service, U.S. Department of Health and Human Services.
Clarkson EM, Warren RL, McDonald SJ, Wardener HE de. 1967. The effect of a high intake of calcium on magnesium metabolism in normal subjects and patients with chronic renal failure. Clin Sci 32:11–18.
Curhan GC, Willett WC, Rimm EB, Stampfer MJ. 1993. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 328:833–838.
Curhan GC, Willett WC, Speizer FE, Spiegelman D, Stampfer MJ. 1997. Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann Intern Med 126:497–504.
Dalton MA, Sargent JD, O’Connor GT, Olmstead EM, Klein RZ. 1997. Calcium and phosphorus supplementation of iron-fortified infant formula: No effect on iron status of healthy full-term infants. Am J Clin Nutr 65:921–6.
Dorsch TR. 1986. The milk-alkali syndrome, vitamin D, and parathyroid hormone. Ann Intern Med 105:800–801.
Ebeling PR, Yergey AL, Vieira NE, Burritt MF, O’Fallon WM, Kumar R, Riggs BL. 1994. Influence of age on effects on endogenous 1,25-dihydroxy-vitamin D on calcium absorption in normal women. Calcif Tissue Int 55:330–334.
French JK, Koldaway IM, Williams LC. 1986. Milk-alkali syndrome following over-the-
Frithz G, Wictorin B, Ronquist G. 1991. Calcium-induced constipation in a prepubescent boy. Acta Paediatr Scand 80:964–965.
Golden BE, Golden MH. 1981. Plasma zinc, rate of weight gain, and the energy cost of tissue deposition in children recovering from severe malnutrition on a cow’s milk or soya protein-based diet. Am J Clin Nutr 34:892–899.
Goldfarb S. 1994. Diet and nephrolithiasis. Ann Rev Med 45:235–243.
Gora ML, Seth SK, Bay WH, Visconti JA. 1989. Milk-alkali syndrome associated with use of chlorothiazide and calcium carbonate. Clin Pharm 8:227–229.
Hakim R, Tolis G, Goltzman D, Meltzer S, Friedman R. 1979. Severe hypercalcemia associated with hydrochlorothiazide and calcium carbonate therapy. Can Med Assoc J 21:591–594.
Hallberg L, Rossander-Hulten L, Brune M, Gleerup A. 1992. Calcium and iron absorption: Mechanism of action and nutritional importance. Eur J Clin Nutr 46:317–327.
Hart M, Windle J, McHale M, Grissom R. 1982. Milk-alkali syndrome and hypercalcemia: A case report. Nebr Med J 67:128–130.
IOM (Institute of Medicine). 1997. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, DC: National Academy Press.
IOM (Institute of Medicine). 1998. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academy Press.
Johnson CM, Wilson DM, O’Fallon WM, Malek RS, Kurland LT. 1979. Renal stone epidemiology: A 25-year study in Rochester, Minn. Kidney Int 16:624–631.
Junor JR, Catto GRD. 1976. Renal biopsy in the milk-alkali syndrome. J Clin Path 29:1074–1076.
Kallmeyer JC, Funston MR. 1983. The milk-alkali syndrome: A case report. S Afr Med J 64:287–288.
Kapsner P, Langsdorf L, Marcus R, Kraemer FB, Hoffman AR. 1986. Milk-alkali syndrome in patients treated with calcium carbonate after cardiac transplantation. Arch Intern Med 146:1965–1968.
Kleinman GE, Rodriquez H, Good MC, Caudle MR. 1991. Hypercalcemic crisis in pregnancy associated with excessive ingestion of calcium carbonate antacid (milk-alkali syndrome): Successful treatment with hemodialysis. Obstet Gynecol 73:496–499.
Kochersberger G, Westlund R, Lyles KW. 1991. The metabolic effects of calcium supplementation in the elderly. J Am Geriatr Soc 39:192–196.
Lemann J Jr, Worcester EM, Gray RW. 1991. Hypercalciuria and stones. Am J Kidney Dis 17:386–391.
Lin S-H, Lin Y-F, Shieh S-D. 1996. Milk-alkali syndrome in an aged patient with osteoporosis and fractures. Nephron 73:496–497.
Malone DNS, Horn DB. 1971. Acute hypercalcemia and renal failure after antacid therapy. Brit Med J 1:709–710.
Massey LK, Roman-Smith H, Sutton RA. 1993. Effect of dietary oxalate and calcium on urinary oxalate and risk of formation of calcium oxalate kidney stones. J Am Diet Assoc 93:901–906.
Matkovic V, Ilich JZ, Andon MB, Hsieh LC, Tzagournis MA, Lagger BJ, Goel PK. 1995. Urinary calcium, sodium, and bone mass of young females. Am J Clin Nutr 62:417–425.
McCarron DA, Morris CD. 1985. Blood pressure response to oral calcium in persons with mild to moderate hypertension: A randomized, double-blind, placebo-controlled, crossover trial. Ann Intern Med 103:825–831.
Moss AJ, Levy AS, Kim I, Park YK. 1989. Use of vitamin and mineral supplements in the United States: Current users, types of products, and nutrients. Advance data from vital and health statistics, No. 174. Hyattsville, MD: National Center for Health Statistics.
Muldowney WP, Mazbar SA. 1996. Rolaids-yogurt syndrome: A 1990s version of milk-alkali syndrome. Am J Kidney Dis 27:270–272.
Newmark K, Nugent P. 1993. Milk-alkali syndrome: A consequence of chronic antacid abuse. Postgrad Med 93:149–156.
Nusser SM, Carriquiry AL, Dodd KW, Fuller WA. 1996. A semiparametric transformation approach to estimating usual daily intake distributions. J Am Stat Assoc 91:1440–1449.
O'Brien KO, Abrams SA, Stuff JE, Liang LK, Welch TR. 1996. Variables related to urinary calcium excretion in young girls. J Pediatr Gastroenterol Nutr 23:8–12.
Orwoll ES. 1982. The milk-alkali syndrome: Current concepts. Ann Intern Med 97:242–248.
Pak CY. 1988. Medical management of nephrolithiasis in Dallas: Update 1987. J Urol 140:461–467.
Recker RR. 1985. Calcium absorption and aclorhydria. N Engl J Med 313:70–73.
Riggs BL, O’Fallon WM, Muse J, O’Conner MK, Melton LJ III. 1996. Long-term effects of calcium supplementation on serum PTH, bone turnover, and bone loss in elderly women. J Bone Miner Res 11:S118.
Robertson, WG. 1985. Dietary factors important in calcium stone formation. In: Schwille PO, Smith LH, Robertson WG, Vahlensieck W, eds. Urolithiasis and Related Clinical Research. New York: Plenum Press. Pp. 61–68.
Sakhaee K, Baker S, Zerwekh J, Poindexter J, Garcia-Hernandez PA, Pak CY. 1994. Limited risk of kidney stone formation during long-term calcium citrate supplementation in nonstone forming subjects. J Urol 152:324–327.
Saunders D, Sillery J, Chapman R. 1988. Effect of calcium carbonate and aluminum hydroxide on human intestinal function. Dig Dis Sci 33:409–412.
Schiller L, Santa Ana C, Sheikh M, Emmett M, Fordtran J. 1989. Effect of the time of administration of calcium acetate on phosphorus binding. N Engl J Med 320:1110–1113.
Schlesinger L, Arevalo M, Arredondo S, Diaz M, Lonnerdal B, Stekel A. 1992. Effect of a zinc-fortified formula on immunocompetence and growth of malnourished infants. Am J Clin Nutr 56:491–498.
Schuman CA, Jones HW III. 1985. The "milk-alkali" syndrome: Two case reports with discussion of pathogenesis. Quart J Med (New Series) 55:119–126.
Shils ME. 1994. Magnesium. In: Shils ME, Olson JA, Shike M, eds. Modern Nutrition in Health and Disease. Philadelphia, PA: Lea & Febiger. Pp. 164–184.
Simmer K, Khanum S, Carlsson L, Thompson RP. 1988. Nutritional rehabilitation in Bangladesh--the importance of zinc. Am J Clin Nutr 47:1036–1040.
Slesinski MJ, Subar AF, Kahle LL. 1996. Dietary intake of fat, fiber, and other nutrients is related to the use of vitamin and mineral supplements in the United States: The 1992 National Health Interview Survey. J Nutr 126:3001–3008.
Smith EL, Gilligan C, Smith PE, Sempos CT. 1989. Calcium supplementation and bone loss in middle-aged women. Am J Clin Nutr 50:833–842.
Snedeker SM, Smith SA, Greger JL. 1982. Effect of dietary calcium and phosphorus levels on the utilization of iron, copper, and zinc by adult males. J Nutr 112:136–143.
Sokoll LJ, Dawson-Hughes B. 1992. Calcium supplementation and plasma ferritin concentrations in premenopausal women. Am J Clin Nutr 56:1045–1048.
Spencer H, Menczel J, Lewin I, Samachson J. 1965. Effect of high phosphorus intake on calcium and phosphorus metabolism in man. J Nutr 86:125–132.
Thys-Jacobs S, Ceccarelli S, Bierman A, Weisman H, Cohen M-A, Alvir J. 1989. Calcium supplementation in prementrual syndrome: A randomized crossover trial. J Gen Intern Med 4:183–189.
Whiting SJ, Wood RJ. 1997. Adverse effects of high-calcium diets in humans. Nutr Rev 55:1–9.
Wood RJ, Zheng JJ. 1990. Milk consumption and zinc retention in postmenopausal women. J Nutr 120:398–403.
B. FOLATE
Hazard Identification
The potential hazards associated with high intake of folate were reviewed as the first step in developing a Tolerable Upper Intake Level (UL). Careful consideration was given to the metabolic interrelationships between folate and vitamin B12, which include (1) shared participation of the two vitamins in an enzymatic reaction; (2) identical hematologic complications resulting from deficiency of either nutrient; (3) amelioration, by folic acid, of the hematologic complications caused by either folate or B12 deficiency; and (4) in B12 deficiency, the occurrence of neurological complications that do not respond to folic acid.
Adverse Effects
No adverse effects have been associated with the consumption of excess folate from foods (Butterworth and Tamura, 1989). Therefore, this review is limited to evidence concerning intake of synthetic folic acid. The experimental data in animal studies and in vitro tissue and cell culture studies were considered briefly to determine if they were supportive of the limited human data.
Neurologic Effects. The risk of neurologic effects described in this section applies to individuals with vitamin B12 deficiency. Vitamin B12 deficiency is often undiagnosed but may affect a substantial percentage of the population, especially older adults. Three types of evidence suggest that excess folic acid intake may precipitate or exacerbate the neurologic damage of B12 deficiency. First, numerous human case reports show onset or progression of neurologic complications in vitamin B12-deficient individuals receiving supplemental folic acid Table IV-3). Second, studies in monkeys (Agamanolis et al., 1976) and fruit bats (van der Westhuyzen and Metz, 1983; van der Westhuyzen et al., 1982) show that vitamin B12-deficient animals receiving supplemental folic acid develop signs of neuropathology earlier than do controls. The monkey studies used dietary methods to induce B12 deficiency, whereas the fruit bat studies used a well-described method involving nitrous oxide (Metz and van der Westhuyzen, 1987). Third, a metabolic interaction between folate and vitamin B12 is well documented (Chanarin et al., 1989). Although the association between folic acid treatment and neurologic damage observed in human case reports does not provide proof of causality, the hazard associated with excess folic acid cannot be ruled out. The hazard remains plausible given the findings from animal studies and the demonstrated biochemical interaction of the two nutrients. The resulting neurologic damage may be serious, irreversible, and crippling.
For many years, it has been recognized that excessive intake of folic acid may obscure or "mask" and potentially delay the diagnosis of vitamin B12 deficiency. Delayed diagnosis can result in an increased risk of progressive, unrecognized neurological damage.
Table IV-3. Dose and Duration of Oral Folic Acid Administration and the Occurrence of Neurologic Manifestations in Patients with Pernicious Anemiaa.
|
Study |
Number of Subjects |
Dose (mg/d) |
Duration |
Occurrence of Neurologic Manifestations b |
|
Crosby, 1960 |
1 |
0.35 |
2 yrs |
1 of 1 |
|
Ellison, 1960 |
1 |
0.33–1 |
3 mo |
1 of 1 |
|
Allen et al., 1990 |
3 |
0.4–1 |
3–18 mo |
3 of 3 |
|
Baldwin and Dalessio, 1961 |
1 |
0.5 |
16 mo |
1 of 1 |
|
Ross et al., 1948 |
4 |
1.25 |
9–23 mo |
1 of 4 |
|
Chodos and Ross, 1951 |
4 |
1.25c |
3.5–26 mo |
3 of 4 |
|
Victor and Lear, 1956 |
2 |
1.5–2.55 |
10–39 mo |
2 of 2 |
|
Conley and Krevans, 1951 |
1 |
4.5 |
3 yr |
1 of 1 |
|
Schwartz et al., 1950 |
48 |
5 |
48 mo |
32 of 48 |
|
Ross et al., 1948 |
2 |
5 |
20–23 mo |
1 of 2 |
|
Conley and Krevans, 1951 |
2 |
5–8 |
2–2.5 yr |
2 of 2 |
|
Will et al., 1959 |
36 |
5–10 |
1–10 yr |
16 of 36 |
|
Bethell and Sturgis, 1948 |
15 |
5–20 |
12 mo |
4 of 15 |
|
Chodos and Ross, 1951 |
11 |
5–30 |
3–25 mo |
7 of 11 |
|
Israels and Wilkinson, 1949 |
20 |
5–40 |
35 mo |
16 of 20 |
|
Wagley, 1948 |
10 |
5–600 |
12 mo |
8 of 10 |
|
Ellison, 1960 |
1 |
5.4–6.4 |
2 yr |
1 of 1 |
|
Victor and Lear, 1956 |
1 |
6.68 |
2.5 yr |
1 of 1 |
|
Berk et al., 1948 |
12 |
10 |
>17 mo |
3 of 12 |
|
Best, 1959 |
1 |
10 |
26 mo |
1 of 1 |
|
Spies and Stone, 1947 |
1 |
10 |
22 d |
1 of 1 |
|
Ross et al., 1948 |
6 |
10–15 |
£ 12 mo |
4 of 6 |
|
Hall and Watkins, 1947 |
14 |
10–15 |
2–5 mo |
3 of 14 |
|
Heinle et al., 1947 |
16 |
10–40 |
£ 12 mo |
2 of 16 |
|
Jacobson et al., 1948 |
1 |
10–65 |
5 mo |
1 of 1 |
|
Heinle and Welch, 1947 |
1 |
10–100 |
4 mo |
1 of 1 |
|
Spies et al., 1948 |
38 |
³ 10 |
24 mo |
28 of 38 |
|
Ross et al., 1948 |
7 |
15 |
28–43 mod |
3 of 7 |
|
Chodos and Ross, 1951 |
1 |
15 |
10.5 mo d |
1 of 1 |
|
Fowler and Hendricks, 1949 |
2 |
15–20 |
4–5 mo |
2 of 2 |
|
Vilter et al., 1947 |
21 |
50–500 |
10–40 d |
4 of 4 |
a
b
Refers to neurologic relapses or progression of preexisting neurologic manifestations while on folic acid therapy.c
In two patients, the neurologic progression was characterized as minimal or slight. Neurologic progression was also observed when the dose was increased to 15 mg/day in these patients.d
The initial dosage of 1.25 mg/day was increased to 15 mg/day after variable durations of treatment. Neurologic progression occurred only at 15 mg/day in these patients.NOTE: Case reports that covered hematologic rather than neurologic effects were excluded, namely, Alperin, 1966; Heinle and Welch, 1947; Herbert, 1963; Reisner and Weiner, 1952; Ritz et al., 1951; Sheehy et al., 1961; Thirkette et al., 1964. All studies except Allen et al. (1990) were conducted before folic acid was added to any foods as a fortificant. In a majority of the case reports for which hematologic status was reported, some degree of hematologic improvement occurred. Studies are presented in increasing order by dose. When different doses were reported within a study, there is more than one entry for that study.
Evidence from animal as well as in vitro tissue and cell culture (Baxter et al., 1973; Hommes and Obbens, 1972; Kehl et al., 1984; Loots et al., 1982; Olney et al., 1981; Spector, 1972; Weller et al., 1994) studies suggests that folic acid is neurotoxic and epileptogenic in animals; however, clear evidence of folic acid-induced neurotoxicity in humans is lacking. Concerns have been raised about the possibility of decreased effectiveness of treatment if individuals treated with anticonvulsants take high doses of folic acid. However, the UL does not apply to drug-drug interactions or to high doses taken under medical supervision.
General Toxicity. In one nonblinded, uncontrolled trial, oral doses of 15 mg of folic acid/day for 1 month were associated with mental changes, sleep disturbances, and gastrointestinal effects (Hunter et al., 1970). However, studies using comparable or higher doses and/or longer durations failed to confirm these findings (Gibberd et al., 1970; Hellstrom, 1971; Richens, 1971; Sheehy, 1973; Suarez et al., 1947).
Reproductive and Developmental Effects. Many studies have evaluated the periconceptional use of supplemental folic acid (in doses of approximately 0.4 to 5.0 mg) to prevent neural tube defects (see Table IV-4). No adverse effects have been demonstrated, but the studies were not specifically designed to assess adverse effects. No reports were found of adverse effects attributable to folic acid among long-term users or among infants born each year to mothers who take supplements, but this has not been investigated systematically. Because it is possible that subtle effects might have been missed, investigations designed to detect adverse effects are needed.
Carcinogenicity. In a large epidemiological study, positive associations were found between folic acid intake and the incidence of cancer of the oropharynx and hypopharynx, and total cancer (Selby et al., 1989). However, the authors of this study suggest that these associations might have been related to unmeasured confounding variables such as alcohol and smoking. Additionally, other studies suggest that folate might be anticarcinogenic (Campbell, 1996).
Hypersensitivity. Individual cases of hypersensitivity reactions to oral and parenteral folic acid have been reported (Gotz and Lauper, 1980; Mathur, 1966; Mitchell et al., 1949; Sesin and Kirschenbaum, 1979; Sparling and Abela, 1985). Such hypersensitivity is rare, but reactions have occurred at folic acid doses as low as 1 mg/day (Sesin and Kirschenbaum, 1979).
Intestinal Zinc Absorption. Although there has been some controversy regarding whether supplemental folic acid intake adversely affects intestinal zinc absorption (Butterworth and Tamura, 1989), a comprehensive review of the literature reveals that folic acid supplementation has either no effect on zinc nutriture or an extremely subtle one (Arnaud et al., 1992; Butterworth et al., 1988; Hambidge et al., 1993; Keating et al., 1987; Milne et al., 1984; Tamura, 1995; Tamura et al., 1992). In a study of prenatal folic acid supplementation, Mukherjee et al. (1984) noted a significant association between the occurrence of fetomaternal complications and the combination of low maternal plasma zinc and high maternal plasma
TABLE IV-4. Assessing Adverse Reproductive Effects from Studies Involving Supplemental Folic Acid
|
Reference |
Subjects |
Duration of Study |
Study Design |
Folate Dose (mg/d) |
Adverse Effects Observed |
Method(s) for Assessing Association and Adverse Effects |
|
Smithells et al., 1981 |
550 women |
110 d (mean duration) |
Clinical trial: controlled |
1 |
None |
NRa |
|
Vergel et al., 1990 |
81 women |
³ 3 mo |
Clinical trial: controlled |
5 |
None |
NR |
|
Czeizel and Dudas, 1992 |
4,753 women (< 35 yr) |
3 mo |
Clinical trial: randomized, controlled |
0.8 |
None |
NR |
|
Laurence et al., 1981 |
95 women |
³ 9 wk |
Clinical trial: randomized, controlled, double-blinded |
4 |
None |
NR |
|
Kirke et al., 1992 |
354 pregnant women |
5 mo |
Clinical trial: randomized, controlled |
0.36 |
None |
NR |
|
Mukherjee et al., 1984 |
450 pregnant women |
³ 9 mo |
Prospective cohort study |
0.4–1b |
Pregnancy complications, fetal distressc |
Statistical association between 12 indices of nutrient status and 7 poorly defined categories of complications |
|
Wald et al., 1991 |
910 women |
A few monthsd |
Clinical trial: randomized, double-blinded, controlled |
4 |
None |
Medical exams performedb |
|
Holmes-Siedle et al., 1992 |
100 women |
Periconceptional period; 7–10 yr follow-up |
Observational study |
1 |
Frequency of developmental anomalies not greater than expectede |
NR |
|
Czeizel et al., 1994 |
5,502 women |
3 mo |
Randomized, controlled trial |
0.8 |
13.4% fetal death rate in supplemented group compared to 11.5% fetal dealth rate on controlsf |
Documentation for all pregnancy outcomes was collected. Statistical evaluation based on two-tailed chi-square test. |
a
b
Plasma folate was measured at different times in pregnancy, but compliance with prenatal vitamin use was not recorded.c
There was no control of confounding variables making it difficult to interpret the results.d
The average duration of exposure is not indicated in the publication, but was likely a few months.e
The frequency of developmental anomalies was not greater than expected; but parental reports of worries, fearfulness, and fussiness among the children were greater than expected.f
This may be a chance finding resulting from multiple comparisons. It has been reported that prenatal multivitamin supplementation (which includes folic acid) can reduce preterm deliveries, causing an apparent increase in recognized abortions as the duration of all pregnancies increases (Scholl et al., 1997).folate concentrations. However, this study may have failed to control for potential confounding factors. Furthermore, these findings are not supported by Tamura and colleagues (1992), who found high serum folate concentrations to be associated with favorable pregnancy outcomes including (1) higher birth weight and Apgar scores of newborns, (2) reduced prevalence of fetal growth retardation, and (3) lower incidence of maternal infection close to the time of delivery.
Summary
The weight of the limited, but suggestive, evidence that excessive folic acid intake may precipitate or exacerbate neuropathy in B12-deficient individuals justifies the selection of this endpoint as the critical endpoint for the development of a UL for folic acid.
Dose-Response Assessment
Adults
Data Selection. To evaluate a dose-response relationship and derive a UL for folic acid, case reports were used that involved oral administration of folic acid in patients with vitamin B12 deficiency who showed development or progression of neurological complications. Because a number of apparently healthy individuals are vitamin B12 deficient (IOM, 1998), these individuals are considered part of the general population in setting a UL.
Identification of a NOAEL or LOAEL. The literature was reviewed to find cases in which B12-deficient patients who were receiving oral folic acid experienced progression of neurologic disorders. Data were not available on which to set a no-observed-adverse effect level (NOAEL). A lowest-observed effect level (LOAEL) of 5 mg of folic acid is based on the data presented in Table IV-3 and summarized below:
1. At doses of folic acid of 5 mg/day and above, there were more than 100 reported cases of neurological progression.
2. At doses of less than 5 mg of folic acid/day (0.33 to 2.5 mg/day), there are only eight well-documented cases.
3. In the majority of cases throughout the dose range, folic acid maintained the patients in hematologic remission over a considerable time span.
4. The background intake of folate from food was not specified, but all except for three cases (those reported by Allen and coworkers [1990]) occurred before the fortification of breakfast cereal with folic acid.
Uncertainty Assessment. An uncertainty factor (UF) of 5 was selected. Compared with the UFs used to date for other nutrients for which there was also a lack of controlled, dose-response data, a UF of 5 is large. The selection of a relatively large UF is based primarily on the severity of the neurological complications observed, but also on the use of a LOAEL rather than a NOAEL to derive the UL. The UF is not larger than 5 based on the uncontrolled observation that millions of people have been exposed to self treatment with about one-tenth of the LOAEL (i.e., 400 m g in vitamin pills) without reported harm.
Derivation of a UL. The LOAEL of 5 mg of folic acid/day was divided by a UF of 5 to obtain the UL for adults as follows:
LOAEL = 5 mg/day = 1 mg or 1,000 T g of folic acid/day
UF 5
A UL of 1,000 m g/day is set for all adults rather than just for the elderly for the following reasons: (1) the devastating and irreversible nature of the neurological consequences, (2) data suggesting that pernicious anemia may develop at a younger age in some racial/ethnic groups (Carmel and Johnson, 1978), and (3) uncertainty about the occurrence of vitamin B12 deficiency in younger age groups. In general, the prevalence of vitamin B12 deficiency among females in the childbearing years is very low, and the consumption of folic acid at or above the UL in this subgroup is unlikely to produce adverse effects.
Folate UL Summary, Adults
UL for Adults Ages 19 years and older 1,000 T g of folic acid/day
Other Life-Stage Groups
There are no data on other life-stage groups that can be used to identify a NOAEL or LOAEL and derive a UL. For infants, the UL was judged not determinable because of lack of data on adverse effects in this age group and concern about the infant’s ability to handle excess amounts. To prevent high levels of intake, the only source of intake for infants should be from food, which would include that provided by fortified products. No data were found to suggest that other life-stage groups have increased susceptibility to adverse effects of high folic acid intake. Therefore, the UL of 1,000 m g/day is also set for adult pregnant and lactating women. The UL of 1,000 m g/day for adults was adjusted for children and adolescents on the basis of relative body weight (see Appendix III) . In some cases, values have been rounded down.
ULs for Children Ages 1–3 years 300 m g of folic acid/day
Ages 4–8 years 400 m g of folic acid/day
Ages 9–13 years 600 m g of folic acid/day
Ages 14–18 years 800 m g of folic acid/day
ULs for Pregnancy Ages 14–18 years 800 m g of folic acid/day
Ages 19 years and older 1,000 m g of folic acid/day
ULs for Lactation Ages 14–18 years 800 m g of folic acid/day
Ages 19 years and older 1,000 m g of folic acid/day
Special Considerations
Individuals who are at risk of vitamin B12 deficiency (e.g., those who eat no animal foods [vegans]) may be at increased risk of the precipitation of neurologic disorders if they consume excess folic acid (IOM, 1998).
Intake Assessment
It is not possible to use data from NHANES III or CSFII to determine the population’s exposure to synthetic folic acid. Currently available survey data do not distinguish between food folate and synthetic folic acid added as a fortificant or taken as a supplement. Based on data from NHANES III and excluding pregnant women (for whom folic acid is often prescribed), the highest reported total folate intake from food and supplements at the ninety-fifth percentile, 983 m g/day, was found in females aged 30 through 50 years. This intake was obtained from food (which probably included fortified ready-to-eat cereal, a few of which contain as much as 400 m g of folic acid/serving) and supplements. For the same group of women, the reported intake at the ninety-fifth percentile from food alone (which also probably included fortified ready-to-eat cereal) was 438 m g/day. In Canada, the contribution of ready-to-eat cereals is expected to be lower because the maximum amount of folic acid that can be added to breakfast cereal is 60 m g/100 g (Health Canada, 1996).
It would be possible to exceed the UL of 1,000 m g/day of synthetic folic acid through the ingestion of fortified foods and/or supplements in typical total diets in the U.S. and Canada (IOM, 1998).
Risk Characterization
The intake of folic acid is currently higher than indicated by NHANES III because enriched cereal grains in the U.S. food supply, to which no folic acid was added previously, are now fortified with 140 m g of folic acid/100 g of cereal grain. Using data from the 1987–1988 U.S. Department of Agriculture’s Nationwide Food Consumption Survey, the U.S. Food and Drug Administration (FDA) estimated that the 95th percentile of folate intakes for males aged 11 to 18 years would be 950 m g of total folate at this level of fortification; this value assumes that these young males would also take supplements containing 400 m g of folic acid (DHHS, 1993). Excluding pregnant women, for whom estimates were not provided, the 95th percentile for total folate for all other groups would be lower, and folic acid intake would be lower still. Using a different method of analysis, the FDA estimated that those who follow the guidance of the Food Guide Pyramid and consume cereal grains at the upper end of the recommended range would obtain an additional 440 m g of folic acid under the new U.S. fortification regulations (DHHS, 1993). (This estimate assumes 8 servings [16 slices] of bread at 40 m g of folic acid per serving and two ~1-cup servings of noodles or pasta at 60 m g of folic acid per serving.) Those who eat other fortified foods (such as cookies, crackers, and donuts) instead of bread might ingest a comparable amount of folic acid. Using either method of analysis and assuming regular use of an over-the-counter supplement that contains folic acid (ordinarily 400 m g per dose), it is unlikely that intake of folic acid would exceed 1,000 m g on a regular basis for any of the life-stage or gender groups.
References
Agamanolis DP, Chester EM, Victor M, Kark JA, Hines JD, Harris JW. 1976. Neuropathology of experimental vitamin B12 deficiency. Neurology 26:905–914.
Allen RH, Stabler SP, Savage DG, and Lindenbaum J. 1990. Diagnosis of cobalamin deficiency. I. Usefulness of serum methylmalonic acid and total homocysteine concentrations. Am J Hematol 34:90–98.
Alperin JB. 1966. Response to varied doses of folic acid and vitamin B12 in megaloblastic anemia. Clin Res 14:52.
Arnaud J, Favier A, Herrmann MA, Pilorget JJ. 1992. Effect of folic acid and folinic acid on zinc absorption. Ann Nutr Metab 36:157–161.
Baldwin JN, Dalessio DJ. 1961. Folic acid therapy and spinal-cord degeneration in pernicious anemia. N Engl J Med 264:1339–1342.
Baxter MG, Millar AA, Webster RA. 1973. Some studies on the convulsant action of folic acid. Br J Pharmacol 48:350–351.
Berk L, Bauer JL, Castle WB. 1948. A report of 12 patients treateed with synthetic pteroylglutamic acid with comments on the pertinent literature. S Afr Med J 22:604–611.
Best CN. 1959. Subacute combined degeneration of spinal cord after extensive resection of ileum in Crohn’s disease: Report of a case. Br Med J 2:862–864.
Bethell FH, Sturgis CC. 1948. The relations of therapy in pernicious anemia to changes in the nervous system. Early and late results in a series of cases observed for periods of not less than ten years, and early results of treatment with folic acid. Blood 3:57–67.
Butterworth CE, Tamura T. 1989. Folic acid safety and toxicity: A brief review. Am J Clin Nutr 50:353–358.
Butterworth CE Jr, Hatch K, Cole P, Sauberlich HE, Tamura T, Cornwell PE, Soong S-J. 1988. Zinc concentration in plasma and erythrocytes of subjects receiving folic acid supplementation. Am J Clin Nutr 47:484–486.
Campbell NR. 1996. How safe are folic acid supplements? Arch Intern Med 156:1638–1644.
Carmel R, Johnson CS. 1978. Racial patterns in pernicious anemia: Early age at onset and increased frequency of intrinsic-factor antibody in black women. N Engl J Med 298:647–650.
Chanarin I, Deacon R, Lumb M, Perry J. 1989. Cobalamin-folate interrelations. Blood Reviews 3:211–215.
Chodos RB, Ross JF. 1951. The effects of combined folic acid and liver extract therapy. Blood 6:1213–1233.
Conley CL, Krevans JR. 1951. Development of neurologic manifestations of pernicious anemia during multivitamin therapy. N Engl J Med 245:529–531.
Crosby WH. 1960. The danger of folic acid in multivitamin preparations. Milit Med 125:233–235.
Czeizel AE, Dudas I. 1992. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med 327:1832–1835.
Czeizel AE, Dudas I, Metneki J. 1994. Pregnancy outcomes in a randomized controlled trial of periconceptional multivitamin supplementation. Final Report. Arch Gynecol Obstet 255:131–139.
DHHS (U.S. Department of Health and Human Services). 1993. Food and Drug Administration. Folic acid; proposed rules. Fed Registr 21:53293–53294.
Ellison ABC. 1960. Pernicious anemia masked by multivitamins containing folic acid. J Am Med Assoc 173:240–243.
Fowler WM, Hendricks AB. 1949. Folic acid and the neurologic manifestations of pernicious anemia. Am Pract 3:609–613.
Gibberd FB, Nicholls A, Dunne JF, Chaput de Saintonge DM. 1970. Toxicity of folic acid. Lancet 1:360–361.
Gotz VP, Lauper RD. 1980. Folic acid hypersensitivity or tartrazine allergy? Am J Hosp Pharm 37:1470–1474.
Hall BE, Watkins CH. 1947. Experience with pteroylglutamic (synthetic folic acid) in the treatment of pernicious anemia. J Lab Clin Med 32:622–634.
Hambidge M, Hackshaw A, Wald N. 1993. Neural tube defects and serum zinc. Br J Obstet Gynecol 100:746–749.
Health Canada. 1996. Departmental Consolidation of the Food and Drugs Act and the Food and Drug Regulations with Ammendments to December 19, 1996. Ottawa: Canada Communications Group.
Heinle RW, Welch AD. 1947. Folic acid in pernicious anemia: Failure to prevent neurologic relapse. J Am Med Assoc 133:739–741.
Heinle RW, Dingle JT, Weisberger AS. 1947. Folic acid in the maintenance of pernicious anemia. J Lab Clin Med 32:970–981.
Hellstrom L. 1971. Lack of toxicity of folic acid given in pharmacological doses to healthy volunteers. Lancet 1:59–61.
Herbert V. 1963. Current concepts in therapy: Megaloblastic anemia. N Engl J Med 268:201–203, 368–371.
Holmes-Siedle M, Lindenbaum RH, Galliard A. 1992. Recurrence of neural tube defect in a group of at risk women: A 10 year study of Pregnavite Forte F. J Med Genet 29:134–135.
Hommes OR, Obbens EA. 1972. The epileptogenic action of Na-folate in the rat. J Neurol Sci 16:271–281.
Hunter R, Barnes J, Oakeley HF, Matthews DM. 1970. Toxicity of folic acid given in pharmacological doses to healthy volunteers. Lancet 1:61–3.
IOM (Institute of Medicine). 1998. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academy Press.
Israels MC, Wilkinson JF. 1949. Risk of neurological complications in pernicious anemia treated with folic acid. Br Med J 2:1072–1075.
Jacobson SD, Berman L, Axelrod AR, Vonder Heide EC. 1948. Folic acid therapy: Its effect as observed in two patients with pernicious anemia and neurologic symptoms. J Am Med Assoc 137:825–827.
Keating JN, Wada L, Stokstad EL, King JC. 1987. Folic acid: Effect of zinc absorption in humans and in the rat. Am J Clin Nutr 46:835–839.
Kehl SJ, McLennan H, Collingridge GL. 1984. Effects of folic and kainic acids on synaptic responses of hippocampal neurones. Neuroscience 11:111–124.
Kirke PN, Daly LE, Elwood JH. 1992. A randomized trial of low-dose folic acid to prevent neural tube defects. Arch Dis Child 67:1442–1446.
Laurence KM, James N, Miller MH, Tennant GB, Campbell H. 1981. Double-blind randomized controlled trial of folate treatment before conception to prevent recurrence of neural tube defects. Br Med J 282:1509–1511.
Loots JM, Kramer S, Brennan MJW. 1982. The effect of folates on the reflex activity in the isolated hemisected frog spinal cord. J Neural Transm 54:239–249.
Mathur BP. 1966. Sensitivity of folic acid: A case report. Indian J Med Sci 20:133–134.
Metz J, Van der Westhuyzen J. 1987. The fruit bat as an experimental model of the neuropathy of cobalamin deficiency. Comp Biochem Physiol 88A:171–177.
Milne DB, Canfield WK, Mahalko JR, Sandstead HH. 1984. Effect of oral folic acid supplements on zinc, copper, and iron absorption and excretion. Am J Clin Nutr 39:535–539.
Mitchell DC, Vilter RW, Vilter CF. 1949. Hypersensivity to folic acid. Ann Intern Med 31:1102–1105.
Mukherjee MD, Sandstead HH, Ratnaparkhi MV, Johnson LK, Milne DB, Stelling HP. 1984. Maternal zinc, iron, folic acid and protein nutriture and outcome of human pregnancy. Am J Clin Nutr 40:496–507.
Olney JW, Fuller TA, de Gubareff T, Labruyere J. 1981. Intrastriatal folic acid mimics the distant but not local brain damaging properties of kainic acid. Neurosci Lett 25:207–210.
Reisner EH Jr, Weiner L. 1952. Studies on mutual effect of suboptimal oral doses of vitamin B12 and folic acid in pernicious anemia. N Engl J Med 247:15–17.
Richens A. 1971. Toxicity of folic acid. Lancet 1:912.
Ritz ND, Meyer LM, Brahin C, Sawitsky A. 1951. Further observations on the oral treatment of pernicious anemia with subminimal doses of folic acid and vitamin B12. Acta Hematol 5:334–338.
Ross JF, Belding H, Paegel BL. 1948. The development and progression of subacute combined degeneration of the spinal cord in patients with pernicious anemia treated with synthetic pteroylglutamic (folic) acid. Blood 3:68–90.
Scholl TO, Hediger ML, Bendich A, Schall JI, Smith WK, Krueger PM. 1997. Use of multivitamin/mineral prenatal supplements: Influence on the outcome of pregnancy. Am J Epidemiol 146:134–141.
Schwartz SO, Kaplan SR, Armstrong BE. 1950. The long-term evaluation of folic acid in the treatment of pernicious anemia. J Lab Clin Med 35:894–898.
Selby JV, Friedman GD, Fireman BH. 1989. Screening prescription drugs for possible carcinogenecity: Eleven to fifteen years of follow-up. Cancer Res 49:5736–5747.
Sesin GP, Kirschenbaum H. 1979. Folic acid hypersensitivity and fever: A case report. Am J Hosp Pharm 36:1565–1567.
Sheehy TW. 1973. Folic acid: Lack of toxicity. Lancet 1:37.
Sheehy TW, Rubini ME, Perez-Santiago E, Santini R Jr, Haddock J. 1961. The effect of "minute" and "titrated" amounts of folic acid on the megaloblastic anemia of tropical sprue. Blood 18:623–636.
Smithells RW, Sheppard S, Schorah CJ, Seller MJ, Nevin NC, Harris R, Read AP, Fielding DW. 1981. Apparent prevention of neural tube defects by periconceptional vitamin supplementation. Arch Dis Child 56:911–918.
Sparling R, Abela M. 1985. Hypersensitivity to folic acid therapy. Clin Lab Haematol 7:184–185.
Spector RG. 1972. Influence of folic acid on exitable tissues. Nature 240:247–249.
Spies TD, Stone RE. 1947. Liver extract, folic acid, and thymine in pernicious anemia and subacute combined degeneration. Lancet 1:174–176.
Spies TD, Stone RE, Lopez GG, Milanes F, Aramburu T, Toca RL. 1948. The association between gastric achlorhydria and subacute combined degeneration of the spinal cord. Postgrad Med 4:89–95.
Suarez RM, Spies TD, Suarez RM Jr. 1947. The use of folic acid in sprue. Ann Intern Med 26:643–677.
Tamura T. 1995. Nutrient interaction of folate and zinc. In: Bailey LB, ed. Folate in Health and Disease. New York: Marcel Dekker. Pp. 287–312.
Tamura T, Goldenberg RL, Freeberg LE, Cliver SP, Cutter GR, Hoffman HJ. 1992. Maternal serum folate and zinc concentrations and their relationships to pregnancy outcome. Am J Clin Nutr 56:365–370.
Thirkette JL, Gough KR, Read AE. 1964. Diagnostic value of small oral doses of folic acid in megaloblastic anemia. Br Med J 1:1286–1289.
van der Westhuyzen J, Metz J. 1983. Tissue S-adenosylmethionine levels in fruit bats with N2O-induced neuropathy. Br J Nutr 50:325–330.
van der Westhuyzen J, Fernandes-Costa F, Metz J. 1982. Cobalamin inactivation by nitrous oxide produces severe neurological impairment in fruit bats: Protection by methionine and aggravation by folates. Life Sci 31:2001–2010.
Vergel RG, Sanchez LR, Heredero BL, Rodriguez PL, Martinez AJ. 1990. Primary prevention of neural tube defects with folic acid supplementation: Cuban experience. Prenat Diagn 10:149–152.
Victor M, Lear AA. 1956. Subacute combined degeneration of the spinal cord. Current concepts of the disease process. Value of serum vitamin B12 determinations in clarifying some of the common clinical problems. Am J Med 20:896–911.
Vilter CF, Vilter RW, Spies TD. 1947. The treatment of pernicious and related anemias with synthetic folic acid. 1. Observations on the maintenance of a normal hematologic status and on the occurrence of combined system disease at the end of one year. J Lab Clin Med 32:262–273.
Wagley PF. 1948. Neurologic disturbances with folic acid therapy. N Engl J Med 238:11–15.
Wald N, Sneddon J, Densem J, Frost C, Stone R. 1991. Prevention of neural tube defects: Results of the Medical Research Council vitamin study. Lancet 338:131–137.
Weller M, Marini AM, Martin B, Paul SM. 1994. The reduced unsubstituted pteroate moiety is required for folate toxicity of cultured cerebellar granule neurons. J Pharmacol Exp Ther 269:393–401.
Will JJ, Mueller JF, Brodine C, Kiely CE, Friedman B, Hawkins VR, Dutra J, Vilter RN. 1959. Folic acid and vitamin B12 in pernicious anemia. Studies on patients treated with these substances over a ten-year period. J Lab Clin Med 53:22–38.
RIBOFLAVIN
Hazard Identification
No adverse effects associated with riboflavin consumption from food or supplements have been reported. Studies involving large doses of riboflavin (Schoenen et al., 1994; Stripp, 1965; Zempleni et al., 1996) have not been designed to systematically evaluate adverse effects. The limited evidence from studies involving large intakes of riboflavin is summarized here.
No adverse effects were reported in humans following single doses up to 60 mg of supplemental riboflavin together with 11.6 mg of riboflavin given intravenously as a single bolus dose (Zempleni et al., 1996). This study is of limited use in setting a Tolerable Upper Intake Level (UL) since it was not designed to assess adverse effects. It is possible that chronic administration of these doses would pose some risk.
In a brief communication, a study by Schoenen and coworkers (1994) stated that no short-term side effects were reported by 48 of 49 patients complaining of migraine heaadachles and treated with 400 mg/day of riboflavin with or without aspirin (75 mg) taken with meals for at least 3 months. Schoenen and coworkers (1994) reported that one patient receiving riboflavin and aspirin withdrew from the study because of gastric upset. This isolated finding is probably an anomaly since no side effects were reported by other patients. Since no clinical or biochemical assessment was undertaken for possible adverse effects, this study by itself is inadequate to use as a basis for determining a NOAEL.
The apparent lack of harm resulting from high oral doses of riboflavin may be due to its limited solubility, humans’ limited capacity to absorb it from the gastrointestinal tract (Levy and Jusko, 1966; Stripp, 1965; Zempleni et al., 1996), and its rapid excretion in the urine (McCormick, 1997). Zempleni et al. (1996) showed that the maximal amount of riboflavin that was absorbed from a single oral dose was 27 mg. A study by Stripp (1965) found limited absorption of 50 to 500 mg of riboflavin with no adverse effects. The poor intestinal absorption of riboflavin is well recognized: riboflavin taken by mouth is sometimes used to mark the stool in experimental studies. There are no data from animal studies suggesting that uptake of riboflavin during pregnancy presents a specific potential hazard for the fetus or infant.
The only evidence of adverse effects associated with riboflavin comes from in vitro studies showing the formation of active oxygen species on intense exposure to visible or ultraviolet (UV) light (Ali et al., 1991; Floersheim, 1994; Spector et al., 1995). However, given the lack of any demonstrated functional or structural adverse effects in humans or animals following excess riboflavin intake, the relevance of this evidence to human health effects in vivo is highly questionable. Nevertheless, it is theoretically plausible that riboflavin increases photo-sensitivity to UV irradiation. Additionally, there is a theoretical risk that excess riboflavin will increase the photosensitized oxidations of cellular compounds such as amino acids and proteins (McCormick, 1977) in infants treated for hyperbilirubinemia, with possible undesirable consequences.
Dose-Response Assessment
The data on adverse effects from high riboflavin intake are not sufficient for a quantitative risk assessment to establish a NOAEL (or LOAEL), and a UL cannot be derived.
Special Considerations
There is some in vitro evidence that riboflavin may interfere with detoxification of chrome VI by reduction to chrome III (Sugiyama et al., 1992). This may be of concern in people who may be exposed to chrome VI; for example, workers in chrome plating. Infants treated for hyperbilirubinemia may also be sensitive to excess riboflavin.
Intake Assessment
Although no UL can be set for riboflavin, an intake assessment is provided here for possible future use. Based on data from NHANES III (unpublished data, R. Briefel, National Center for Health Statistics, Centers for Disease Control and Prevention, 1997), the highest mean intake of riboflavin from diet and supplements for any life-stage and gender group was reported for males aged 31 through 50 years: 6.9 mg/day. The highest reported intake at the ninety-fifth percentile was 11 mg/day in females over age 70 years.
Risk Characterization
No adverse effects have been associated with excess intake of riboflavin from food or supplements. This does not mean that there is no potential for adverse effects resulting from high intakes. Since data on the adverse effects of riboflavin intake are limited, caution may be warranted.
References
Ali N, Upreti RK, Srivastava LP, Misra RB, Joshi PC, Kidwai AM. 1991. Membrane damaging potential of photosensitized riboflavin. Indian J Exp Biol 29:818–822.
Floersheim GL. 1994. Allopurinol, indomethacin and riboflavin enhance radiation lethality in mice. Pediatrics 139:240–247.
McCormick DB. 1977. Interactions of flavins with amino acid residues: assessments from spectral and photochemical studies. Photochem Photobiol 26:169–182.
McCormick DB. 1997. Riboflavin. In: Shils ME, Olson JE, Shike M, Ross AC, eds. Modern Nutrition in Health and Disease. Baltimore, MD: Williams and Wilkins.
Levy G, Jusko WJ. 1966. Factors affecting the absorption of riboflavin in man. J Pharm Sci 55:285–289.
Schoenen J, Lenaerts M, Bastings E. 1994. Rapid communication: High-dose riboflavin as a prophylactic treatment of migraine: Results of an open pilot study. Cephalalgia 14:328–329.
Spector A, Wang GM, Wang RR, Li WC, Kleiman NJ. 1995. A brief photochemically induced oxidative insult causes irreverisible lens damage and cataracts. 2. Mechanism of action. Exp Eye Res 60:483–493.
Stripp B. 1965. Intestinal absorption of riboflavin by man. Acta Pharmacol Toxicol 22:353–362.
Sugiyama M, Tsuzuki K, Lin X, Costa M. 1992. Potentiation of sodium chromate (VI)-induced chromosomal aberrations and mutation by vitamin B2 in Chinese hamster V79 cells. Mutat Res 283:211–214.
Zempleni J, Galloway JR, McCormick DB. 1996. Pharmacokinetics of orally and intravenously administered riboflavin in healthy humans. Am J Clin Nutr 63:54–66.